with Autosomal Dominant Polycystic Kidney Disease (ADPKD) is unique, as
patients typically witness the course of the disease in their affected family
members. This is often a parent, but due to the complexities of life including difficult
family dynamics, estrangement, or early fatality from other causes (ie. trauma,
cancer, cardiovascular disease), it may also be a sibling, parent’s siblings,
or a grandparent. At first assessment, they commonly ask “Am I going to end up like
them?” Contrarily, up to 15% of patients present with no family history (PMID:
28522688), and the delivery of an ADPKD diagnosis and prognosis can be
devastating and seemingly out of the blue.
recently, many nephrologists felt there was no therapeutic options to offer
patients with ADPKD, resulting in therapeutic nihilism until patients reached
advanced stages of chronic kidney disease. Following TEMPO (PMID: 23121377) and
REPRISE (PMID: 29105594), which reported significant reductions in the rate of
GFR decline and kidney growth but with significant cost and side effects, and
awaiting an FDA decision projected for April 2018, nephrologists will increasingly
need to decide whether “the juice is worth the squeeze”.
medicine aims to identify the best patient for a particular treatment using information
from patient history, examination, and bloodwork, as well as incorporating more
advanced imaging, genetics, and biomarkers, as well as patient preferences and
values. Providing an accurate prognosis to patients is important for selecting
the right interventions, but also for life planning for the patient and resource
planning for the system.
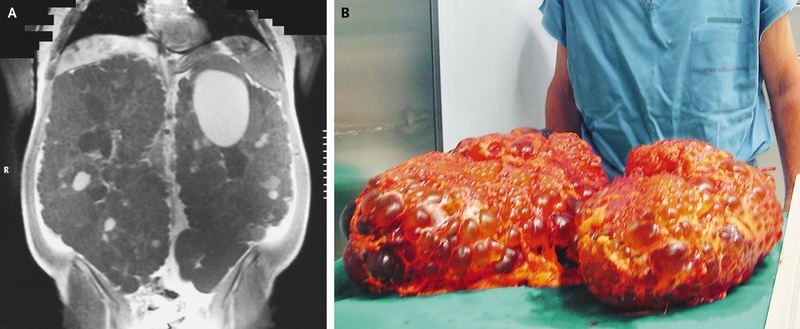
Diagnosis
can be made with ultrasound, especially in those with family history and age
over 30, which has allowed time for cysts to grow. Assessment with Magnetic
Resonance Imaging (MRI) including calculation of age and height adjusted Total
Kidney Volume (http://www.mayo.edu/research/documents/pkd-center-adpkd-classification/doc-20094754)
is the best method for ADPKD risk stratification (PMID: 24904092) and has been
approved by the FDA as a prognostic enrichment biomarker for clinical trial design
(https://www.fda.gov/downloads/Drugs/Guidances/UCM458483.pdf). However, the terms of use include
a specific disclaimer against use in clinical care. Furthermore, the economic
ramifications of MRI screening of ADPKD patients needs to be considered.
Genetic
testing is increasingly available, and while costs are falling and our
knowledge of the prognostic implications of mutation type is increasing,
recommendations remain against widespread testing of all patients (PMID:
25786098). Cases where genetic testing may be of particular importance include:
cases with unclear diagnosis (ie. loss of kidney function without enlargement);
cases without family history; severe early onset disease; families with
intrafamilial discordance; atypical lopsided or unilateral appearance; suspicion
of another syndromic presentation (ie. nephronopthosis, autosomal dominant tubulointerstitial
kidney disease, tuberous sclerosis) or exclusion of disease in a young patient by
checking for a known familial mutation. Nonetheless, it is now recognized that
patients with truncating PKD1
mutations (large deletions, nonsense, frameshift, and canonical splice site
mutations) have the worst prognosis, followed by PKD1 non-truncating mutations (including inframe
insertions/deletions and missense mutations), and PKD2 mutations (PMID: 26453610). After exhaustive screens, those
without mutations detected tend to have the mildest disease. However, due to a
high degree of allelic heterogeneity, determining if a rare mutation is in fact
causal of ADPKD in a specific patient remains far from trivial and
interpretation of sequencing results requires specific training and experience.
is required to specifically identify who needs what testing (imaging, genetics,
or both), and what the specific benefits are of obtaining either or both types
of information. It appears quite likely that those with the greatest risk of
disease progression towards kidney failure have the most to gain from disease
modifying therapies, especially if they are associated with a therapeutic
burden. Future predictive techniques will incorporate imaging and genetics and our
growing knowledge of the cyst physiology and polycystin-1 and polycystin-2, and
utilize tools such as artificial intelligence, to improve precision medicine
care of patients with ADPKD.
Matthew
Lanktree @MattLanktree
Heritable Kidney Disease Post Doctoral Fellow
University
Health Network
University of Toronto
