Welcome to the 41st case of the Skeleton Key Group, a team of nephrologists from around the world who build a periodic education package for the Renal Fellow Network.
Authors : Ali Rizvi, DO, Rajeev Sachdeva, MD
Mentor: Amy Yau, MD
A. The Stem
A 45-year-old woman with end-stage liver disease secondary to alcohol use and multiple recent admissions for hepatic encephalopathy presents with abdominal pain, diarrhea, and altered mental status. The patient was recently discharged home after treatment of decompensated cirrhosis but due to financial factors, was unable to take rifaximin and lactulose as prescribed.
Vitals:
Blood pressure: 119/78 mm Hg
Pulse: 74 bpm
Respiration rate: 14 per minute
Temperature: 97.8 F
Physical Exam:
Constitutional: ill-appearing
HEENT: Normocephalic and atraumatic, dry mucus membranes, scleral icterus present bilaterally
Cardiovascular: Normal rate and regular rhythm. Palpable peripheral pulses.
Pulmonary: No respiratory distress, no wheezing, rales or rhonchi.
Abdominal: Soft without any fluid wave, no focal tenderness noted. No hepatosplenomegaly.
Musculoskeletal: Normal range of motion. No neck rigidity noted. Trace non-pitting edema noted of bilateral lower extremities. No joint deformities or swelling.
Skin: Warm and dry. Jaundiced.
Neurological: Patient is awake with eyes open, able to track movement in the room. However she is confused and not providing verbal responses to questioning. Does not follow commands with her extremities, but extremities do withdraw to noxious stimuli.
B. The Labs
Blood
Magnesium: 2.3 mg/dL
Calcium: 8.7 mg/dL
Albumin: 2.6 g/dL
Corrected Calcium: 9.8 mg/dL
Ammonia: 163 umol/L
Lactic acid 1.1 mmol/L
Hepatic panel:
Total protein 5.6 g/dL
Alkaline phosphatase 252 U/L
AST 160 U/L
ALT 57 U/L
Total Bilirubin 26.5 mg/dL
Direct bilirubin 20.59 mg/dL
WBC 23.9k
Venous Blood Gas:
pH 7.39/ pCO2 22/ pO2 69/ HCO3 12.9
Urine:
Spot studies:
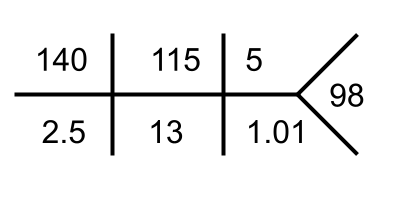
Urine Na 61 mmol/L
Urine K 77 mmol/L
Urine Urea 197 mg/dL
Urine Osmolality 374 mOsm/kg
All 24-hour studies:
Total volume 687 cc
Urine pH 6.3
Sodium 21 mmol
Potassium 61 mmol
Chloride 31 mmol
Calcium 300 mg
Ammonium 18 mmol
Creatinine 0.9 g
Urinalysis: Dark yellow, trace blood, 3+ bilirubin, Spec Grav 1.023, pH 7.5, 10-20 RBCs, 10-20 WBCs, trace bacteria, Specific Gravity 1.023
C. The Workup
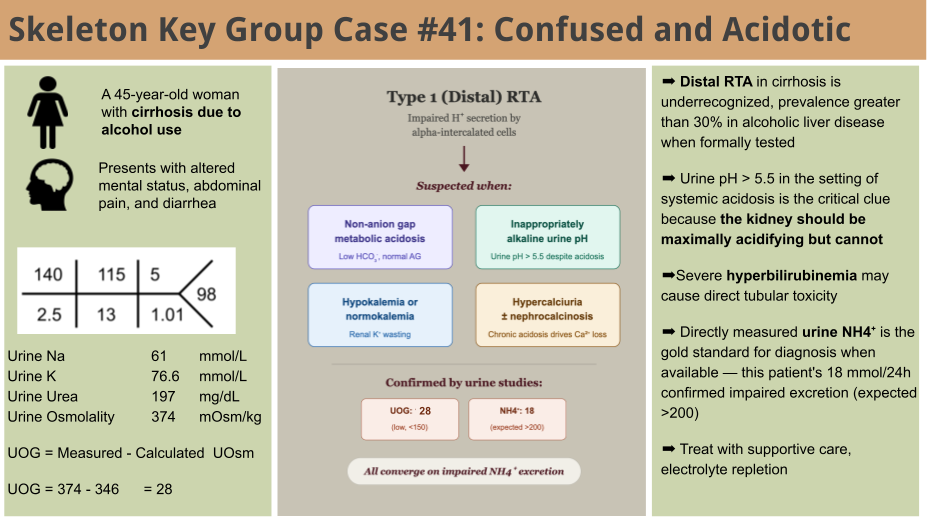
The acid-base profile drew attention. Rather than a single disorder, a stepwise read reveals three coexisting processes hiding behind a nearly normal pH.
Step 1. Identify the primary disorder. The venous blood gas showed a pH of 7.39 with a bicarbonate of 12.9 mEq/L and an appropriately low pCO2 of 22 mmHg = a primary metabolic acidosis.
Step 2. Calculate the anion gap, then correct for albumin. The serum anion gap is Na – Cl – HCO3 = 140 – 115 – 13 = 12, which appears normal. But the albumin is only 2.6 g/dL, and each 1 g/dL below 4.0 lowers the expected anion gap by roughly 2.5. The albumin-corrected anion gap is elevated at 15 (12 + 2.5 × 1.4). In a patient with active alcohol use, poor oral intake, and a normal lactate (1.1 mmol/L), this mild anion-gap acidosis could be explained by ketoacidosis.
Step 3. Evaluate for a second metabolic process (delta ratio). Look for a second metabolic process (delta ratio). ΔAG / ΔHCO3 = (15.5 − 12) / (24 − 13) = 3.5 / 11 = 0.3. A ratio well below 1 means the bicarbonate has fallen much more than the anion gap has risen, pointing to a coexisting non-anion-gap metabolic acidosis (NAGMA).
Step 4. Assess respiratory response. Winter’s formula predicts pCO2 = (1.5 × 13) + 8 = 27.5 ± 2 mmHg. The measured pCO2 of 22 mmHg falls below this range, indicating a superimposed respiratory alkalosis which is a common finding in cirrhosis.
So three processes coexist behind a near-normal pH: a mild AGMA (ketoacidosis), the dominant NAGMA (distal RTA), and a respiratory alkalosis.
In a patient with diarrhea, GI bicarbonate losses would be the expected explanation. But the urine pH of 7.5 was a clue. An inappropriately alkaline urine suggests the kidney itself cannot excrete acid pointing toward a renal tubular acidosis. Urine ammonium > 200 mmol/day is expected to be an adequate response to an acid load. Blunted values are implicated in RTA. Here we measured urine ammonia. But because urine ammonium is not always measured directly, the urine anion gap (UAG) or urine osmolar gap (UOG) can be used as a quick surrogate. The important point is that UAG and UOG are surrogates for the urinary ammonium response, and they are only interpretable in a patient who is already acidemic. Both answer one narrow question: given systemic acidosis, is the kidney mounting an appropriate ammonium response? Outside of acidosis the question itself is not meaningful.
(A quick note on the difference between the patients urine urinalysis pH 7.5 and the 24-hour collection pH 6.3, a study found that dipstick pH was inaccurate in 25% of cases!)
A Urine osmolar gap (UOG) was calculated as: UOG = Measured Osmolality – Calculated Urine Osmolality
The urine studies showed:
Urine Na 61 mmol/L
Urine K 77 mmol/L
Urine Urea 197 mg/dL
Urine Osmolality 374 mOsm/kg
In this case the calculation goes as follows:
Urine Osmolality = 2 x (Urine Na + Urine K) + (Urine Urea/2.8) = 2 x (61+77) + 197/2.8 = 275 + 70 = 345.
UOG = 374 – 346
UOG = 28
An important note on UAG and UOG: Of note, the urine anion gap (UAG = Urine Na + Urine K − Urine Cl) was introduced by Batlle et al. specifically for the evaluation of hyperchloremic (non-gap) metabolic acidosis. In that setting, a positive UAG implies low ammonium excretion (a renal cause, e.g., distal RTA), while a negative UAG implies a robust ammonium response (e.g., GI bicarbonate loss). The UAG works because chloride is excreted alongside ammonium; when ammonium rises, urine chloride rises and the gap turns negative. The UAG has several limitations. It assumes ammonium is excreted as ammonium chloride. When other unmeasured anions accompany ammonium like ketoacids (β-hydroxybutyrate, acetoacetate) the UAG can stay falsely positive (or less negative) and underestimate ammonium. High dietary potassium distorts it. Potassium from plant-based diets arrives largely with organic anions and phosphate rather than chloride, raising urine K in the equation without a matching rise in Cl and inflating the gap independently of ammonium. The urine osmolar gap (UOG = Measured − Calculated urine osmolality) is the more robust surrogate within acidosis because it is less sensitive to variation in measured electrolytes and to unmeasured anions. Finally, the gold standard is direct 24-hour urine ammonium measurement, which has become increasingly available and removes the assumptions underlying both surrogates. In this case the directly measured value (18 mmol/24h) is definitively low and settles the question. This patient demonstrated a decreased urine osmolar gap (< 150) indicating no (or minimal) urinary ammonium consistent with the 24 urine ammonium level .
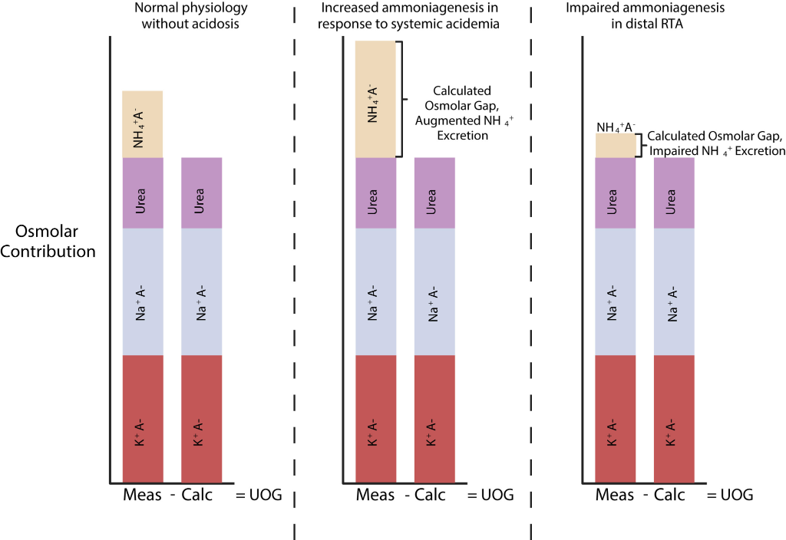
Figure from Bonner et al. The urine osmolar gap in states of appropriate ammonium excretion and ineffective ammonium excretion. Abbreviations: K – potassium, A – anion, Calc – calculated osmolarity, Meas – measured osmolarity, Na – sodium, NH4+ – ammonium, RTA – renal tubular acidosis, UOG – urine osmolar gap.
D. The Diagnosis
This is a 45-year-old woman with end-stage liver disease secondary to alcohol use disorder presents with altered mental status, generalized weakness, abdominal pain, and diarrhea. Initial workup revealed a normal-anion gap metabolic acidosis (pH 7.39, HCO₃⁻ 12.9 mEq/L), hypokalemia, and an elevated ammonia (163 umol/L).While her diarrhea could account for GI bicarbonate losses, the urine pH of 7.5 was inappropriately alkaline. In the setting of systemic acidosis, the kidneys should maximally acidify the urine to a pH <5.5. This was the critical clue suggesting a renal acidification defect. Subsequent 24-hour urine studies confirmed markedly low urinary ammonium excretion (18 mmol/24h; expected >300 mmol/day), with a corroborating decreased urine osmolal gap (28). Taken together with the hypokalemia, the findings are suggestive of a Type 1 (distal) renal tubular acidosis in the setting of end-stage liver disease.
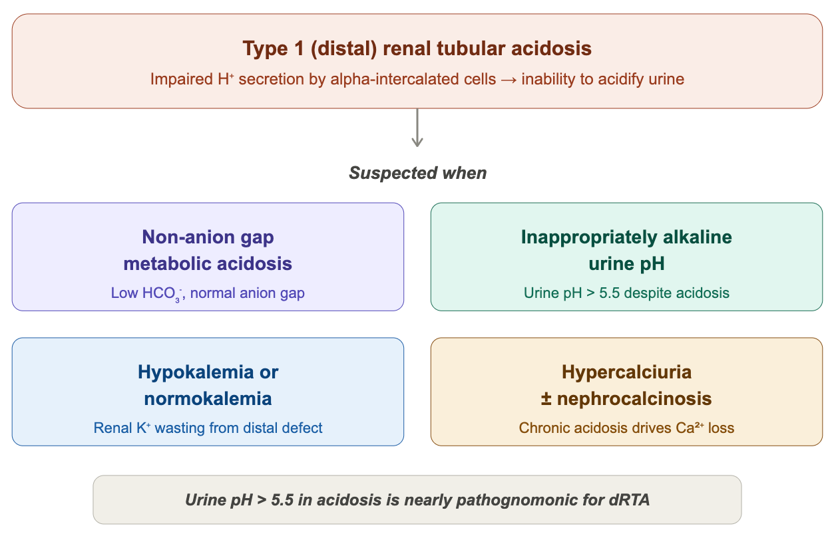
Figure: Diagnosis of Type 1 RTA
Differential Diagnosis of Non-Anion Gap Metabolic Acidosis
Skeleton Key Tip: Urine pH >5.5 during systemic acidosis is a clue for distal (Type 1) RTA. Please note: using urine pH>5.5 as an absolute criterion for distal RTA has its own caveats, such as a patient with urinary tract infection which can produce urease and lead to alkaline urine, and hence not always considered pathognomonic.
What is renal tubular acidosis (RTA) and how do we clinically approach these patients?
Renal tubular acidosis is part of a spectrum of diseases that contribute to normal anion gap metabolic acidosis (NAGMA). NAGMA is primarily due to renal vs extrarenal (GI) losses of hydrogen. RTA is the development of acidosis either due to impaired bicarbonate reabsorption or insufficient ammonium (NH4+) excretion.
Pulmonary and renal systems work together to maintain acid-base homeostasis. One of the lungs primary responsibilities is to remove ~15,000 mmol CO2 (volatile carbonic acid) that is generated from carbohydrate and fat metabolism on a daily basis. The kidneys, on the other hand, take care of excreting nonvolatile acids. In normal physiologic conditions, the kidney excretes 1 mmol/kg/day on nonvolatile acids that are byproducts of sulfur-containing amino acid metabolism (mainly methionine and cysteine). Remaining free hydrogen ions need a buffer in order to be efficiently excreted by the kidneys, namely dibasic phosphate and ammonia. The development of RTA hinges around the elevated level of these buffers in the urine, and primarily via NH4+ production. NH4+ is synthesized in the proximal tubular cells by deamination of glutamine to glutamate and NH4+. NH4+ gets absorbed into the interstitium in the thick ascending limb by the NKCC2 co-transporter by taking the place of potassium via molecular mimicry. Potassium and NH4+ share near-identical ionic radii and hydration energies, leading to the transporter being “tricked” into treating them interchangeably. NH4+ then dissociates to ammonia (NH3). NH3 diffuses into the collecting duct and gets ‘trapped’ as NH4+ in acidic urine. Critically, for every NH4+ excreted in the urine, a new bicarbonate molecule is generated and returned to the blood. This is why urinary ammonium excretion in systemic acidosis far exceeds the baseline nonvolatile acid load of ~1 mmol/kg/day. The kidney is not only disposing of daily acid production but regenerating the bicarbonate stores that have been consumed by the acidosis. In the presence of systemic acidosis, without any kidney defect in urinary acidification, this adaptive response drives urinary ammonium excretion to >200 mmol/day when needed. In RTA, this response is blunted and small urinary ammonium excretion values (< 40 mmol/day) are seen.
Why does Type 1 RTA occur in liver disease?
The association between distal RTA and cirrhosis has long been recognized. In a1969 NEJM study, Shear et al. demonstrated impaired urinary acidification in 9 of 15 patients with cirrhosis after acid loading, with accompanying defects in renal potassium conservation. In 1981 Caregaro et al. found that RTA occured in 33% of patients with cirrhosis, with the highest incidence in alcoholic use disorder specifically. In liver disease associated with alcohol use, the prevalence of incomplete distal RTA may be as high as 41% when formally tested with an acid load (Oster et al., 1975), and those patients tended to have higher serum bilirubin levels and worse coagulopathy.
The pathogenesis is likely multifactorial. In autoimmune liver diseases, the mechanism may be related to immune-mediated interstitial nephritis. In alcohol-associated cirrhosis, the mechanisms are less clearly defined but severe hyperbilirubinemia as seen in this patient with a total bilirubin of 27 mg/dL can cause direct tubular toxicity through bile cast nephropathy (cholaemic nephrosis), in which conjugated bilirubin and bile salts deposit in distal tubules and collecting ducts, causing damage and impaired acidification. Volume depletion and the avid proximal sodium reabsorption characteristic of cirrhosis may limit distal sodium delivery, reducing the electrochemical gradient needed for H+ secretion. Finally, alcohol itself may have direct nephrotoxic effects on tubular function independent of liver disease.
Clinical pearl: Distal RTA may be unmasked or exacerbated in patients with volume depletion, hypokalemia, or nutritional deficiencies, all common in patients with liver failure . The coexistence of RTA and cirrhosis is likely underrecognized, and formal acid-base evaluation should be considered in patients with cirrhosis with unexplained hypokalemia or persistent NAGMA.
Management Principles of Type 1 RTA in Liver Disease
The patient was managed with intravenous fluids, aggressive potassium repletion, thiamine supplementation, and nutritional support with refeeding precautions. She was stabilized for ongoing management of her liver failure.
E. Teaching Points
- Type 1 RTA can present subtly in adults, often unmasked by illness, malnutrition, or liver disease.
- Alkaline urine in the setting of metabolic acidosis is a key diagnostic towards a type 1 RTA.
- The coexistence of RTA and cirrhosis is likely underrecognized, and formal acid-base evaluation should be considered in cirrhotic patients with unexplained hypokalemia or persistent NAGMA.
- Electrolyte repletion and alkali therapy are cornerstone therapies; correct underlying issues to prevent recurrence.
Editors: Payal Gaggar, Jefferson Triozzi, Carolina Mendez, Joel Topf, Shweta Shah, Matthew A. Sparks
Reviewed by: Srinath Yadlapalli, M.D.