
In the week that Steve Jobs stepped down as CEO of Apple, it seems appropriate to tip our cap to that guru of simplicity, and take a stripped down, simplified approach to one of the biggest quagmires in nephrology, membranoproliferative glomerulonephritis (MPGN). The existing classification of this entity is a shambles, and it understandably had Emily reaching for the bottle when she recently attempted to get her head around it. To make matters worse, we probably need to throw out all the old literature on immunosuppressive treatment for MPGN and start afresh, as the existing trials were conducted at a time when the full heterogeneity of MPGN was not appreciated. Few evidence-based treatment recommendations are possible based on these data. Well, help is at hand, in the form of a practical and comprehensive new classification. My thanks to Dr. Sanjeev Sethi, MD, PhD at the Mayo Clinic for sending me a copy to review.
My main gripe with the existing classification system is that it places the focus firmly on “primary” causes of MPGN (Types I, II, III, Burkholder variant, Strife and Anders variant etc.), demoting secondary causes to the bottom of the differential. Although idiopathic forms undoubtedly exist, they are increasingly uncommon, and an underlying cause can be found in the vast majority of cases.
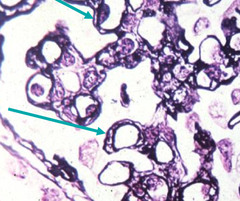
First off, it is vital to appreciate that MPGN is not a disease, but a pattern of injury with a broad differential. For a nephrologist to say their patient “has MPGN” just doesn’t cut it, no more than an ID doc saying their patient “has infection”. The classic presentation is a child or young adult with persistent proteinuria, often with overlapping features of the nephrotic and nephritic syndromes, and depressed C3 (occurs in over 80%). Spontaneous remission is rare and the condition typically grumbles on to ESRD over many years, unless a treatable underlying cause can be found. The hallmark biopsy finding is the double contour on light microscopy (adjacent figure). The double contour may be thought of as the fingerprint of chronic and persistent immunoglobulin/complement-mediated endothelial injury. Endothelial and mesangial cells attempt to repair the ongoing injury by generating basement membrane–like material, which traps immune complexes and cellular elements in between to form double contours.
The proposed classification system is similar to that used for rapidly progressive or crescentic GN, where classification by immunoflourescence pattern on the the renal biopsy into linear, granular and pauci immune staining is extremely helpful in narrowing down the equally broad and complex differential. The proposed MPGN classification divides it into 3 IF pattern groups: Immunoglobulin staining with C3, C3 staining without immunoglobulin and no staining (figure below; modified version of that which appears in article).
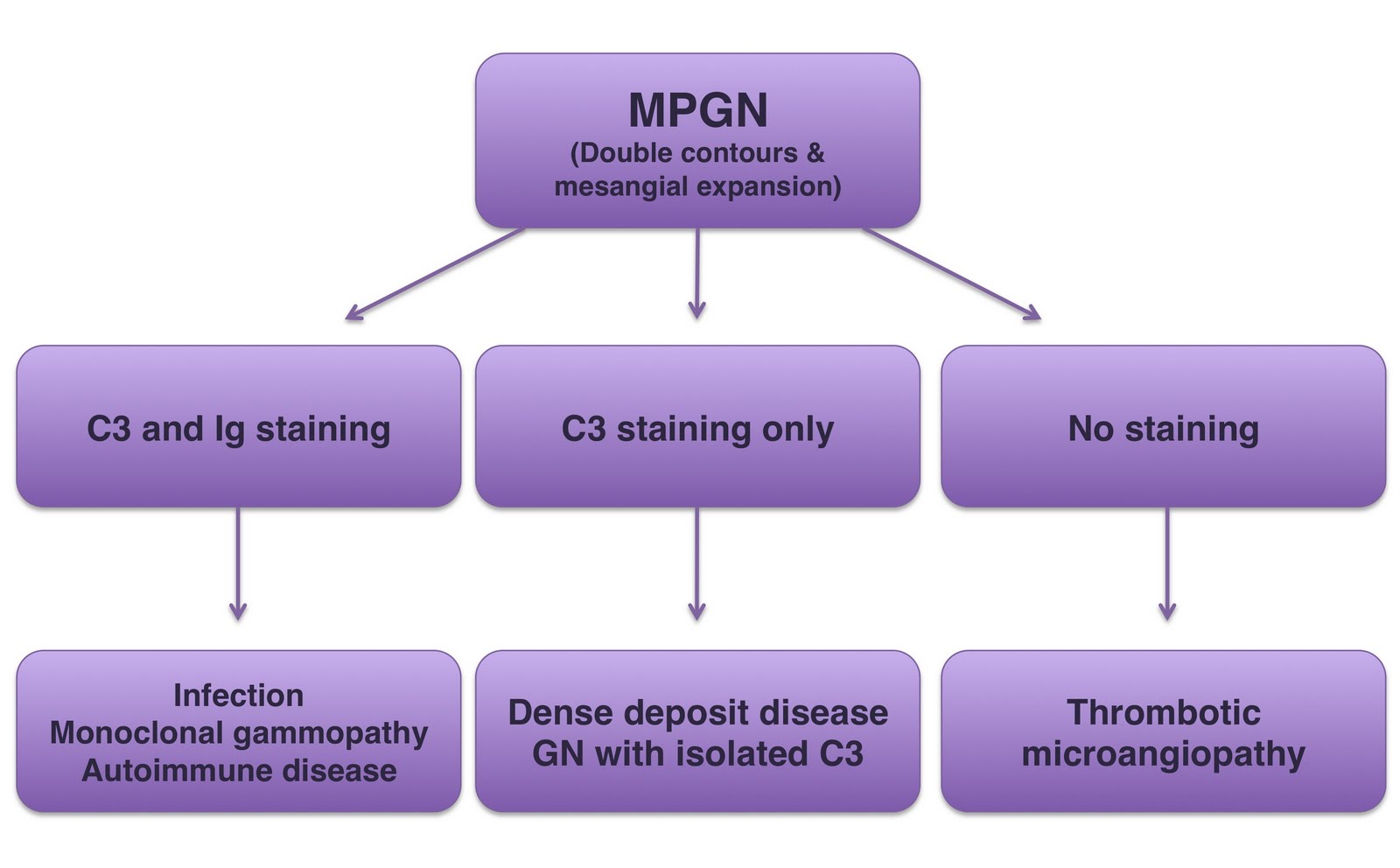
Immunoglobulin and C3 +ve: If immunoglobulin staining is present, the patient should be worked-up for infection, monoclonal gammopathy and autoimmune disease. Regarding infective causes, the organisms to think about tend to be classically associated with chronic infection (HCV, HBV, TB, coxiella, brucella, nocardia) or a carrier state (mycoplasma, Neisseria, strep), presumably acting as a source of chronic, low-grade antibody production. When infective and autoimmune causes are excluded, an underlying MGUS is found in over 40% of cases (Mayo clinic series). This is best identified by immunofixation, although IF microscopy on the biopsy specimen using monoclonal protein–specific Abs is often very helpful. Less common associations include myeloma, lymphoma and CLL.
C3 +ve and immunoglobulin -ve: Isolated C3 staining (i.e.without immunoglobulin) suggests an abnormality of the alternative complement pathway. When this pattern occurs in children, it suggests a genetic mutation is responsible, whereas complement autoantibodies are more likely to be responsible in adults. The prototypic diseases are dense deposit disease and MPGN with isolated C3 deposits, however there are many others. A detailed interrogation of the alternative complement pathway will probably require an immunologist’s input. However, before calling them, a good initial screen of the alternative complement pathway comprises 4 tests: C3 and C4 levels, serum MAC levels, an alternative pathway functional assay (APFA) and hemolytic complement assays.
No staining: Chronic injury to the endothelial cell due to thrombotic microangiopathy also causes an MPGN pattern. However, staining for immunoglobulin and C3 is absent. Causes to consider include TTP, HUS, anti-phospholipid antibody syndrome, drug-associated TMA, nephropathy associated with bone marrow transplantation, radiation nephritis and malignant hypertension.
Hopefully, using this system, you will be able to quickly hone in on a manageable differential diagnosis, leaving you more time to… think.


http://www.nephronpower.com/2010/07/clinical-case-19-answer-and-summary.html
Check out a quiz I had created a while back that homes in the point you are trying to make.
Kenar