Cardiac and renal involvement usually manifest after the first or second decades of life. Fabry’s nephropathy initially presents as proteinuria or isosthenuria, later progressing to renal insufficiency. By age 35 years, half of male Fabry’s patients have proteinuria, and by age 50, about half have developed ESRD. While some female heterozygotes develop renal insufficiency, they do so less frequently, and rarely progress to end-stage disease.
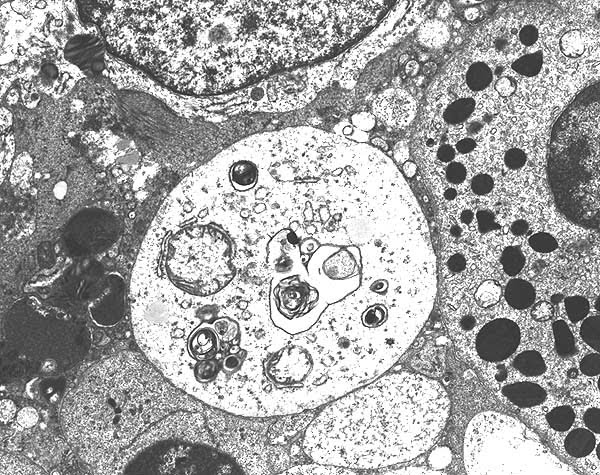
Biopsy findings in Fabry disease show lysosomal inclusion bodies containing glycolipid material in visceral epithelial cells, most prominently podocytes. “Zebra bodies”, inclusions of ceramide material in lysosomes, are often seen in the podocytes. Vascular sclerosis along with other chronic changes such as interstitial fibrosis and tubular atrophy are also commonly seen. Fabry’s can be diagnosed definitively by various serum tests measuring the alpha-galactosidase A activity. Sequencing of the GLA gene, mutations of which are known to cause Fabry’s disease, can be helpful in assessing female family members for presence of a carrier state.
Currently, two formulations of recombinant purified alpha-galactosidase A exist, although only one (Fabrazyme, alpha-galactosidase A alpha) is on the market in the U.S. While trials have not been effective in reversing renal damage in ESRD patients, they have shown a significant slowing in the rate of EGFR decrease in patients with mild renal dysfunction (EGFR > 55 mL/min). Despite the inability to rescue kidney function in our dialysis patients, however, screening plays a vital function in establishing family inheritance patterns—and identifying the next generation of Fabry’s patients in the early stages of disease. So, in younger male patients with ESRD of unclear etiology: ask about Fabry’s symptoms, screen them, and help to increase Fabry’s awareness and diagnosis!
Originally posted by Lisa Cohen

