Abdul Qader, MD
NSMC 2021
Department of Pediatric Nephrology
Square Hospitals Ltd
Dhaka, Bangladesh
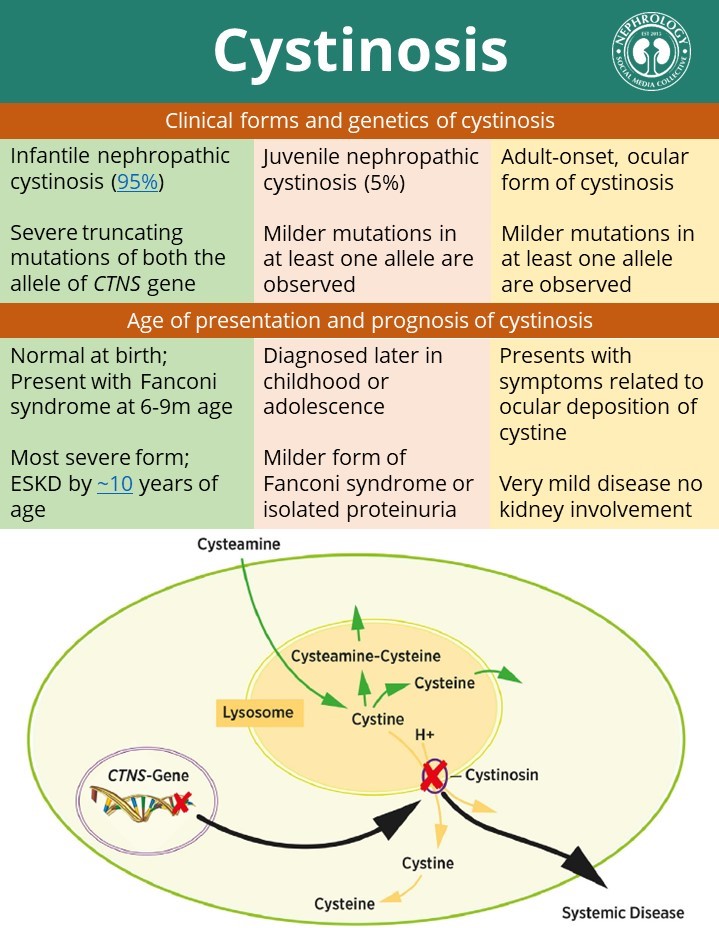
Introduction: Cystinosis is the most common cause of childhood renal Fanconi syndrome. It is a lysosomal storage disorder characterized by the accumulation of cystine in the lysosomes of multiple organs. Cystinosis is inherited in an autosomal recessive manner and CTNS (17p 13.2) is the responsible gene for this disorder. CTNS encodes cystinosin, which is a carrier protein for lysosomal cystine transport. In this essay, we’ll go over the basics of cystinosis, how we treat it, and what the future holds. We’ll also try to figure out what we don’t know about cystinosis.
What do we know about cystinosis:
Epidemiology: The incidence of cystinosis in the general population is about 1 in 100,000 to 200,000 live births. A higher incidence was reported in the French Brittany (1 in 2600 live births) and Saguenay- Quebec (1 in 62500 live births) due to distinct mutations in CTNS gene (c.898-900+24del127 and p.Trp138X respectively). However, precise data on prevalence are lacking. According to NAPRTCS (North American Pediatric Renal Trials and Collaborative Studies) 2011 and 2014 data, cystinosis comprises 1.4% of children on dialysis and 2.1% among the pediatric kidney transplant recipients in the United States (US).
Pathogenesis: Cystine accumulates in the proximal tubular cell (PTC) and interstitial macrophage in two forms either as soluble cystine or cystine crystal. A soluble cystine, which can organize into polyhedral crystals. Crystals were found in the proximal tubule cells of CTNS mutant mice’s kidneys, whereas crystals were found primarily in the interstitial macrophages of children. CTNS knockout mice also showed evidence of Fanconi syndrome long before crystal formation. Crystal formation likely causes fibrosis and leads to progressive kidney failure. Cystine crystal deposition begins in the tubuloglomerular junction and progresses to the proximal tubule, causing proximal tubular cells apoptosis. The thick columnar epithelial cells of proximal tubules are then replaced by the flat epithelium of the bowman’s capsule by expansion into the tubule (sliding metaplasia). This epithelial metaplasia would lead to the formation of swan neck deformity. There is evidence that decreased expression of apical endocytic receptors, megalin, and cubilin in the proximal tubular cell surfaces of CTNS knockout mice. Megalin and cubilin are responsible for the reuptake of lysosomal degradation of protein, so that could explain the low molecular proteinuria that develops with Fanconi syndrome.
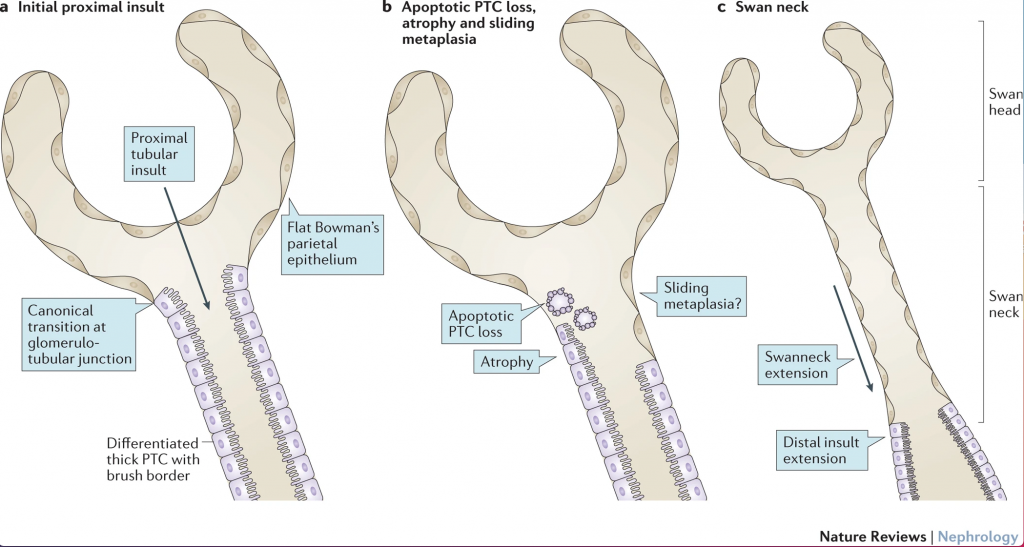
There is a lower abundance of adenosine triphosphate (ATP) in the tubular cells in cystinosis compared to normal tubular cells likely due to altered cellular energy homeostasis. Cystine is the main source of cysteine which is used for the synthesis of glutathione in the cytosol. So, reduced glutathione, which is the major cytosolic antioxidant, along with neutralization of free radicals like oxygen species have been proposed as the mechanism for Fanconi syndrome. On the other hand, cystinosin plays a vital role in the lysosomal exocytosis, autophagy, and oxidative state of the sensing cells. Cysteamine used for effective cystine depletion can decrease apoptotic cell death and cell oxidation in vivo but is unable to reverse the effect of Fanconi syndrome.
Clinical presentations:
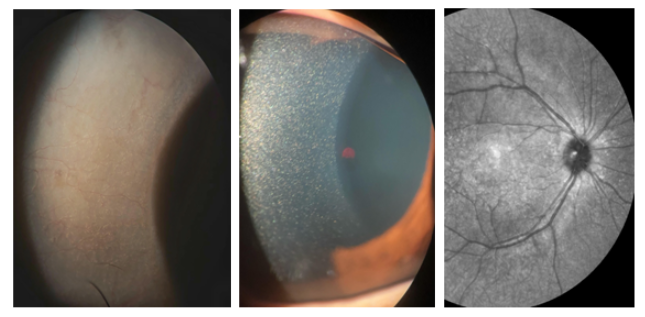
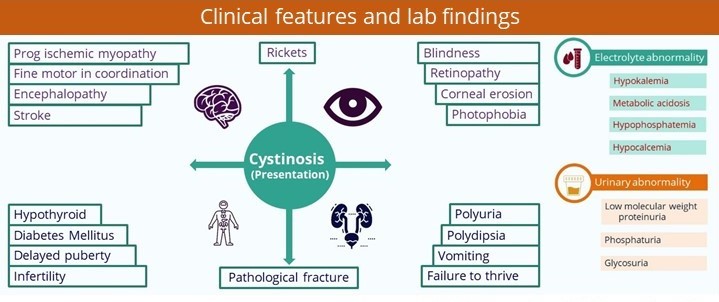
Diagnosis:
A diagnosis of nephropathic cystinosis can be confirmed with the following tests:
- Measurement of leukocyte cystine level (LCL), Level >2 nmol half cystine/mg protein is diagnostic. (Normal: <0.2 nmol half cystine/mg protein)
- Presence of corneal cystine in slit lamp exam
- Genetic analysis of CTNS gene.
Prenatal diagnosis is also possible on DNA samples from chorionic villi or from amniotic fluid cells.
Treatment:
- Dietary recommendations: Children should receive 100% of the recommended dietary allowance (RDA) of calories for their age. The composition should be balanced and include salt and fluid supplements. To prevent growth failure, early placement of gastric tubes as well as jejunal tubes for very young children who have a poor appetite and frequent vomiting.
- Therapy with cysteamine: Cysteamine was introduced for human use in the 1990s and it effectively reduces LCL levels and delays progression to kidney failure. Cysteamine enters the lysosome via amino thiol transporter and cleaves cystine to cysteine or cysteine-cysteamine mixed disulfide cysteine. This leads cysteine-cysteamine to exit from the lysosome via lysine carrier.
- Cysteamine bitartrate (Cystagon or Procysbi), which is given orally for the treatment of cystinosis.
- Cystagon is taken orally every 6 hours. A target dose is 1.3 mg/m2/day of the free base (for patients up to 12 years of age) and 2 g/day for patients older than 12 years old or weighing >50 kg. The starting dose should be one-sixth of the target dose and gradually increase the dose over 4-6 weeks. The maximum dose should not be exceeded 1.95 mg/m2/day.
- Procysbi) is taken orally every 12 hours and a starting dose for cysteamine naive patients is 0.2 to 0.3 g/m2/day.
- Topical cysteamine hydrochloride eye drops are used for dissolving the crystals and for symptom relief. A 0.44% cysteamine ophthalmic solution (Cystaran) and 0.55% cysteamine gel (Cystadrops) can be used.
- Adverse drug reactions of cysteamine include foul odor, diarrhea, gastrointestinal upset, and headache, which makes it difficult to achieve compliance.
- Price of the drugs: Cystagon 50mg capsule- € 93 (100 capsules); 150mg capsule- €239 (100 capsules); Cystadrops® 5ml- €295 per bottle.
- Cysteamine bitartrate (Cystagon or Procysbi), which is given orally for the treatment of cystinosis.
- Other therapies:
- Indomethacin: Indomethacin is used orally at a dose of 1-3 mg/kg/day in 2-3 divided doses. It decreases the expression of aquaporin 2 thus enhances salt reabsorption and improves polyuria and clinical symptoms. But the chronic indomethacin toxicity is a particular concern.
- Renin-angiotensin system blockade: Angiotensin-converting enzyme inhibitor (ACEi) and or angiotensin receptor blocker (ARBs) can decrease albuminuria and delay the progression of renal function but lack of evidence for systemic use in patients with nephropathic cystinosis as the children are already have compromised kidney function due to severe salt and water loses secondary to Fanconi syndrome.
- Therapies for Fanconi syndrome:
- Correction of electrolyte imbalance including alkalinization with citrate (Polycitra, Bicitra, or Potassium citrate). Hypokalemia and hyponatremia correction.
- Prevention of rickets with replacement of phosphate and vitamin D supplement as required.
- Therapies for endocrine abnormalities:
- Hormone replacement in hypothyroidism and growth hormone therapy.
- Insulin for treatment of diabetes mellitus.
- Testosterone therapy for male hypogonadism.
- Kidney transplantation: Kidney transplantation is the treatment of choice for children with kidney failure. The Proximal tubular disorder does not recur in transplanted kidneys but the metabolic abnormalities related to Fanconi syndrome may persist if the native kidney continues to produce urine. Overall the result of kidney transplantation is better in cystinosis patients compared to the other patients.
Potential new targets:
- Hematopoietic stem cell transplantation: In irradiated CTNS knockout mice, either purified mesenchymal stem cells (MSC) or hematopoietic stem cells (HSC) were transplanted to repopulate their bone marrow with CTNS carrying donor cells. The outcome showed, MSC provides short-term protection but HSC leads to a long-term improvement in the kidney structure and Fanconi syndrome.
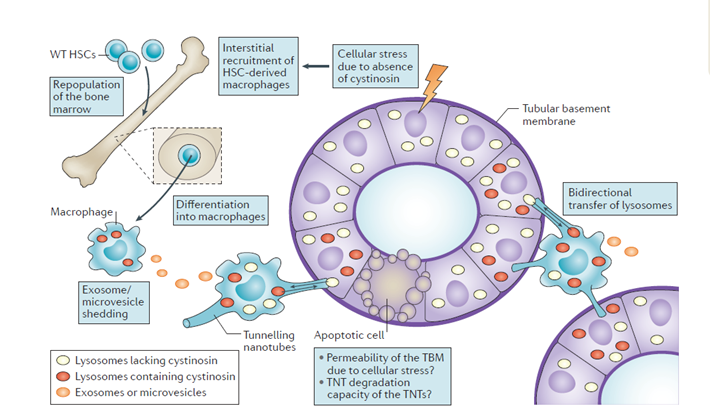
The proposed mechanism is bone marrow-derived macrophages engrafted in cystinotic kidneys generate nanotubular extension that readily crosses the tubular basement membrane and deliver cystinosin into diseased proximal tubule cells. Similar protection is also observed in the eyes and thyroid after HSC.
- Genistein: Genistein is an angiogenesis inhibitor and a phytoestrogen. This compound is found in soy products like soybeans and soy milk. Genistein can activate TFEB in PTC of cystinotic kidney and can decrease intracellular cysteine level and lysosome enlargement and rescue endocytosis defect. Genistein has previously been reported to decrease lysosomal storage of glycosaminoglycans in mucopolysaccharidosis in vivo and has been validated in clinical trials showing improvement of cognition.
- ELX- 02: ELX- 02 is a eukaryotic ribosomal selective glycoside (ERSG), which enables the production of a sufficient amount of functional protein to restore activity. ELX- 02 was found effective in reducing white cell cystine level and completed phase I trial and the phase II trial is currently suspended because of revision of study design to include patients older than 12 years.
- Current clinical trials for stem cell transplantation: Phase 1/2 Clinical trial including adolescent and adult (>14 years), where they will receive hematopoietic stem cell transplantation of gene-modified own stem cell (ex vivo gene- modified with a lentiviral vector, pCCL- CTNS to express CTNS gene product name: CTNS-RD-04).
Outcomes:
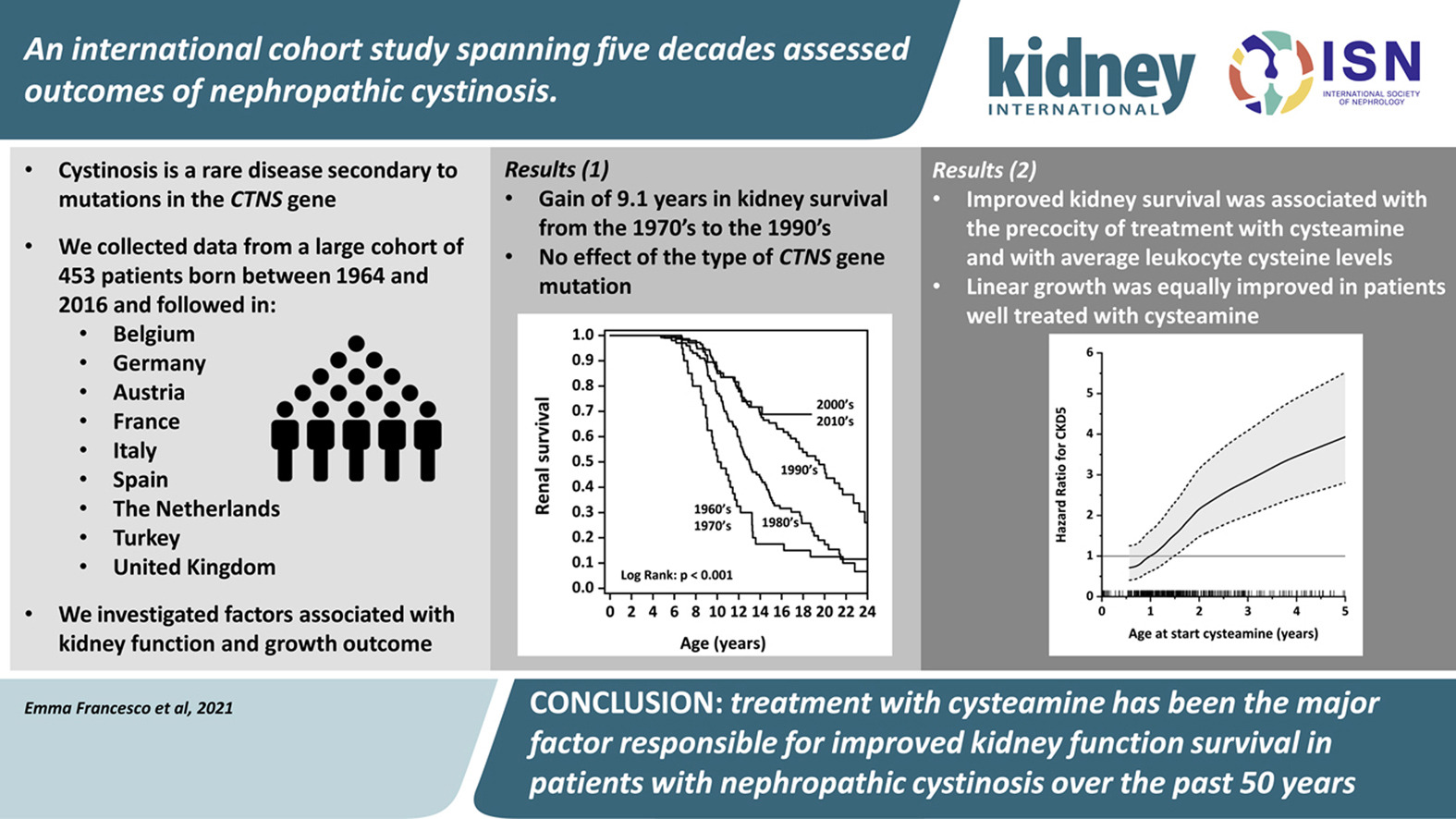
In a recent multicentre study by Emma et al. reported the outcome of 453 nephropathic cystinosis and showed cysteamine has a major influence on improving kidney function and delaying progression to ESKD.
Areas of uncertainty and potential research targets:
With the recent advancements of research, several aspects in the pathogenesis and therapeutic goal have come up and created uncertain areas in cystinosis patient management.
The role of cystine crystals in the pathogenesis of interstitial fibrosis is still unknown. Moreover, with the treatment of cysteamine, the changes in the proximal tubular cells are irreversible and recent research revealed there is an abnormal mTORC1 activation which also contributes to the PTC damage. So, details of the pathogenic mechanisms and therapeutic options for these are still unknown. The role of early administration of cysteamine from birth in preventing renal Fanconi syndrome is still unknown.
Some children show severe proteinuria without renal Fanconi syndrome. So details of podocyte involvements in cystinosis are still unknown. Regarding treatments options, adverse events profile and compliance to cysteamine are bringing up the necessity of therapeutic alternatives to this medication. The toxicity related to lifetime use of cysteamine is unknown and use during pregnancy and lactation is still debatable. The timing of sperm harvesting and preservation for future use in male cystinosis patients is still unknown.
Conclusion: Cystinosis is an autosomal recessive lysosomal storage disorder due to mutation in the CTNS gene. Treatment with cysteamine has been shown to improve kidney function and decrease multisystem complications as well as delay progression to kidney failure but does not reverse changes in the proximal tubule cells. Newer agents like Genistein and ELX-02 are promising in decreasing intracellular cystine level which is currently under trials. A recent study on CTNS knockout mice showed HSC transplantation can improve proximal tubule cell changes & kidney function, which is currently under trial in patients older than 14 years.
Reviewed by Matthew A Sparks, S. Sudha Mannemuddhu, Swasti Chaturvedi, Amy Yau

