Sami Droubi, MD, Raphael Rosen, MD
Fellows, New York Presbyterian / Columbia University Medical Center
For decades it has been suspected that a “permeability factor” is present in the serum of patients with idiopathic nephrotic syndrome but the identity of this factor has eluded investigators.
In the 1950s, serum from infants with nephrotic syndrome was injected into normal infants and caused a transient increase in proteinuria, suggesting that something in the infant serum was causative. This was again replicated (somewhat more ethically) by injecting serum patients with recurrent FSGS into rat glomerudli, which also caused glomerular permeability to protein.
In 1972, Hoyer et al described 3 cases of idiopathic nephrotic syndrome that recurred in the allograft and noted that an unknown “circulating humoral substance” in the recipients may be causing this recurrence. Multiple daring reports have shown that in patients with early FSGS recurrence in the allograft, the kidney can be explanted and then transplanted into a non-FSGS recipient with resolution of nephrotic syndrome and adequate graft function. This further implies that simply removing the normal kidney from the abnormal “permeability factor” in the recipient serum cured the impending FSGS.
Complicating this question is the possibility that there could be different permeability factors in minimal change disease vs FSGS or whether there could be multiple culprit factors in each.
Investigators proposed, in a paper published in JASN, the newest candidate for a permeability factor in minimal change disease, anti-nephrin antibodies.
Hypothesis: The investigators noted that Congenital Nephrotic Syndrome of the Finnish (CNF), a rare hereditary form of minimal change disease, occurs due to a mutation in nephrin, a key component of slit diaphragm. They postulate that autoantibodies against nephrin could be the cause of acquired minimal change disease.
Anti-nephrin antibodies in patients with minimal change disease vs controls: First, investigators wanted to determine whether anti-nephrin antibodies are detectable during active disease. An indirect ELISA assay was developed against nephrin and the threshold for positivity was chosen to be 187 U/mL, the maximum titer detected in a healthy control population. Next, investigators assayed the serum of patients from the Nephrotic Syndrome Study Network (NEPTUNE) cohort who had biopsy-confirmed minimal change disease. Antibody levels above 187 U/mL were present in 18 of 62 (29%) patients. Only one of 54 patients (2%) with anti-PLA2R positive membranous nephropathy tested positive for anti-nephrin antibodies.
Eleven of the 18 patients in the NEPTUNE cohort who were anti-nephrin antibody positive during active disease had a subsequent serum sample available during remission. Anti-nephrin antibodies were reduced or completely absent during remission.

But minimal change disease is negative on immunofluorescence … right? Usually, minimal change disease, as the name suggests, is normal on light microscopy and shows negative staining on immunofluorescence, with its real abnormality only detectable on electron microscopy with the finding of diffuse podocyte foot process effacement. So how can these investigators claim that immunoglobulins against nephrin are causing this disease if there is no immunoglobulin detected on most biopsies?
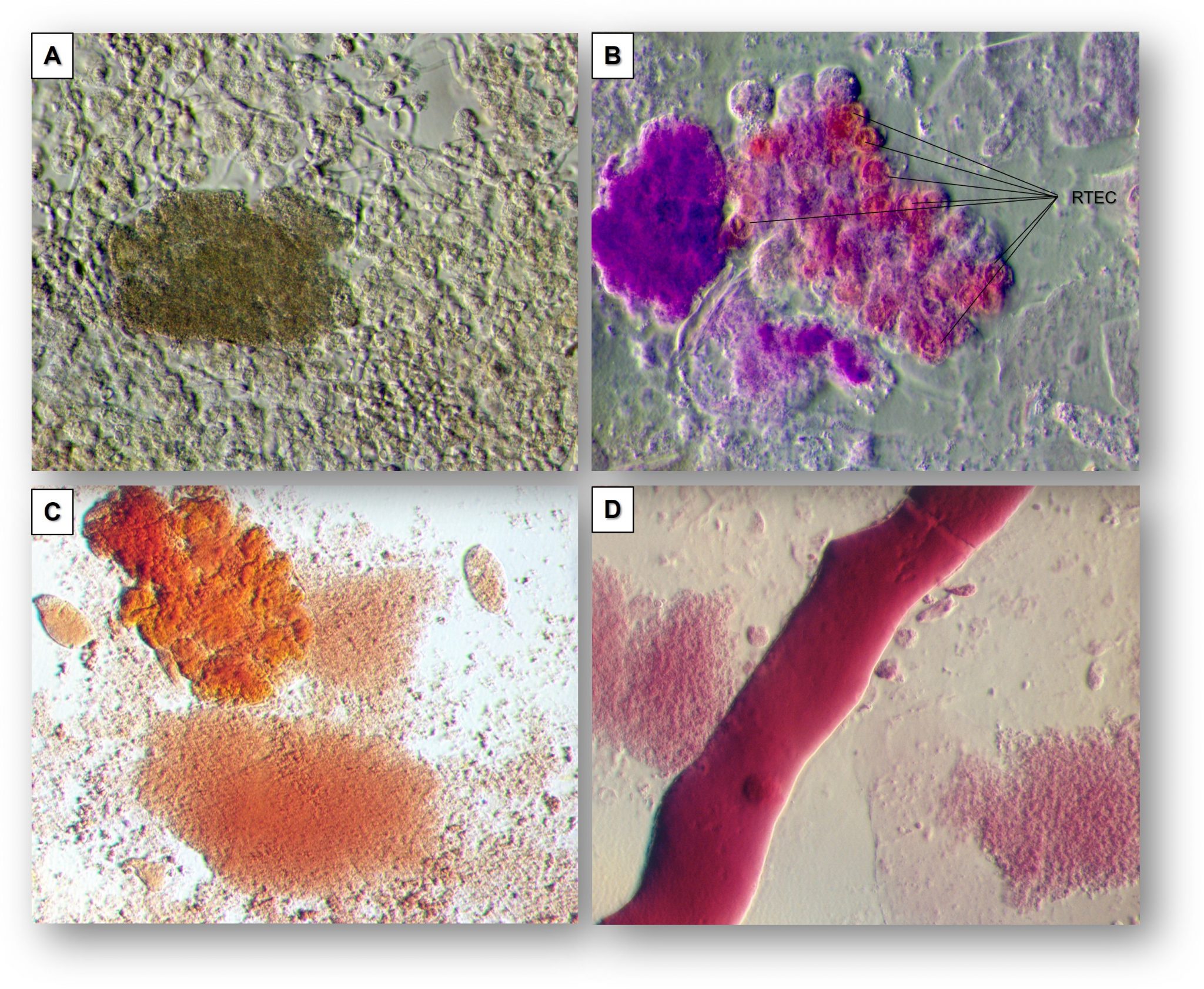
It turns out that the generalization that minimal change disease stains negatively for IgG is only partially true. For years, it has been recognized that some biopsies actually have subtle IgG staining, though certainly not nearly as impressive as the robust staining in membranous nephropathy. These investigators combed through biopsies at their hospitals for ones that had this previously neglected subtle dusting pattern of IgG.

They demonstrated that these IgGs are colocalized with nephrin but not with the podocyte cytoskeletal protein synaptopodin. Remarkably, the patients with this positive punctate IgG staining all had positive serum anti-nephrin antibodies, in contrast to those with negative IgG stain on biopsy, who were uniformly serologically negative.
Finally, the authors describe a case to highlight the possible clinical use of this discovery. A 27-year-old woman with no genetic basis for nephrotic syndrome developed steroid-dependent minimal change disease in childhood which then progressed to FSGS and then kidney failure. She was found to have high levels of anti-nephrin antibodies prior to transplantation. After transplant she developed massive proteinuria. She was subsequently treated with rituximab and plasmapheresis, with elimination of anti-nephrin antibodies and resolution of nephrotic syndrome.
Can we definitively say that anti-nephrin antibodies are the cause of minimal change disease in these patients? The most recent prior candidate for the permeability factor in FSGS was soluble urokinase plasminogen activator receptor (suPAR). When this was ultimately shown not to be the causative factor, the disappointment that followed led some to establish criteria that must be met before a putative permeability factor can be considered causative.
Let us see if these criteria have been met.
| Criteria | Anti-nephrin | Verdict |
| Permeability factor must have activity in vitro and in vivo and be confirmed in validation studies . | Anti-nephrin antibodies have been demonstrated to cause proteinuria in vivo | ✅ |
| The permeability factor must be present in well-characterized affected patients (idiopathic nephrotic syndrome) but not in appropriate controls (i.e. normal patients or other glomerular disease) | Anti-nephrin antibodies are present in a subset of minimal change disease patients at much higher levels than normal controls or PLA2R positive membranous nephropathy. | ✅ |
| There must be temporal plausibility between the permeability factor and disease (i.e. its presence precedes disease phenotype, elevated levels correlate with disease relapse and resolve with disease remission) | In the subset of patients with minimal change disease with elevated anti-nephrin Abs, the levels of anti-nephrin Abs were much higher during disease relapse than during disease remission. However, we do not yet have evidence that development of these antibodies preceded initial onset of the disease. | ✅ ⛔ |
| Specific removal or blocking of the permeability factor results in improvement in symptoms (i.e. not just en-masse removal of antibodies with plasmapheresis – rather, specific targeting of this factor) | This study does not present data that the specific blocking of anti-nephrin antibody causes remission of minimal change disease (PLEX and anti-CD20 therapies reduce all antibody levels, so cannot be said to be causing efficacy due to inhibition of anti-nephrin ab specifically) | ⛔ |
Tell it to me straight – Is the discovery of anti-nephrin antibodies in minimal change disease the equivalent of the anti-PLA2R discovery in membranous nephropathy?
Yes…
The discovery of anti-PLA2R antibodies in the glomeruli and serum of most primary membranous nephropathy patients quickly led to therapeutic monitoring of serum PLA2R levels in determination of disease activity. The discovery of anti-nephrin antibody “dusting” in these biopsy specimens is accompanied by the presence of high levels of anti-nephrin antibody in the serum. It is tempting to fantasize about a future in which a non-invasive diagnosis can be made of anti-nephrin minimal change disease, as has become commonplace with PLA2R-positive membranous nephropathy.
…and no
Despite the subsequent discovery of a myriad of rarer autoantigens in membranous nephropathy, anti-PLA2R remains the causative antibody in the vast majority of primary membranous nephropathy. In this study, even in an enriched cohort of biopsy-proven minimal change disease without pathogenic genetic variants, only 29% were positive for anti-nephrin antibodies. If anti-nephrin antibodies only explain a small subsection of minimal change disease, the impact of this discovery will be much less than that of PLA2R. However, the authors note that nearly all the patients in the NEPTUNE cohort had initiated therapy prior to collection of serum for analysis, which may have decreased their native titers of anti-nephrin antibodies. It is possible that a much higher proportion of patients with new-onset / treatment-naive minimal change disease may have anti-nephrin antibodies.
Future directions: An exciting prospect is whether the anti-nephrin antibody is not only causative in minimal change disease but may also be the driver behind some cases of primary FSGS. At the most recent ASN Kidney Week, two posters (one from the same group at this JASN study and one from a different group were presented that demonstrated anti-nephrin staining in FSGS (PO1675, PO1676).
Reviewed by Matthew A. Sparks

Thank you. Updated. This was supposed to say electron not light.
Excellent post. I just want to point out the foot process effacement in MCD is seen on EM not light as described in post.