Welcome to the 27th case of the Skeleton Key Group, a team of 50-odd nephrology fellows who work together to build a monthly education package for the Renal Fellow Network. These are actual cases (without patient identifying information) that intrigued the treating physician.
Written by : Mythri Shankar
Visual Abstract by : Mythri Shankar
A. The Stem
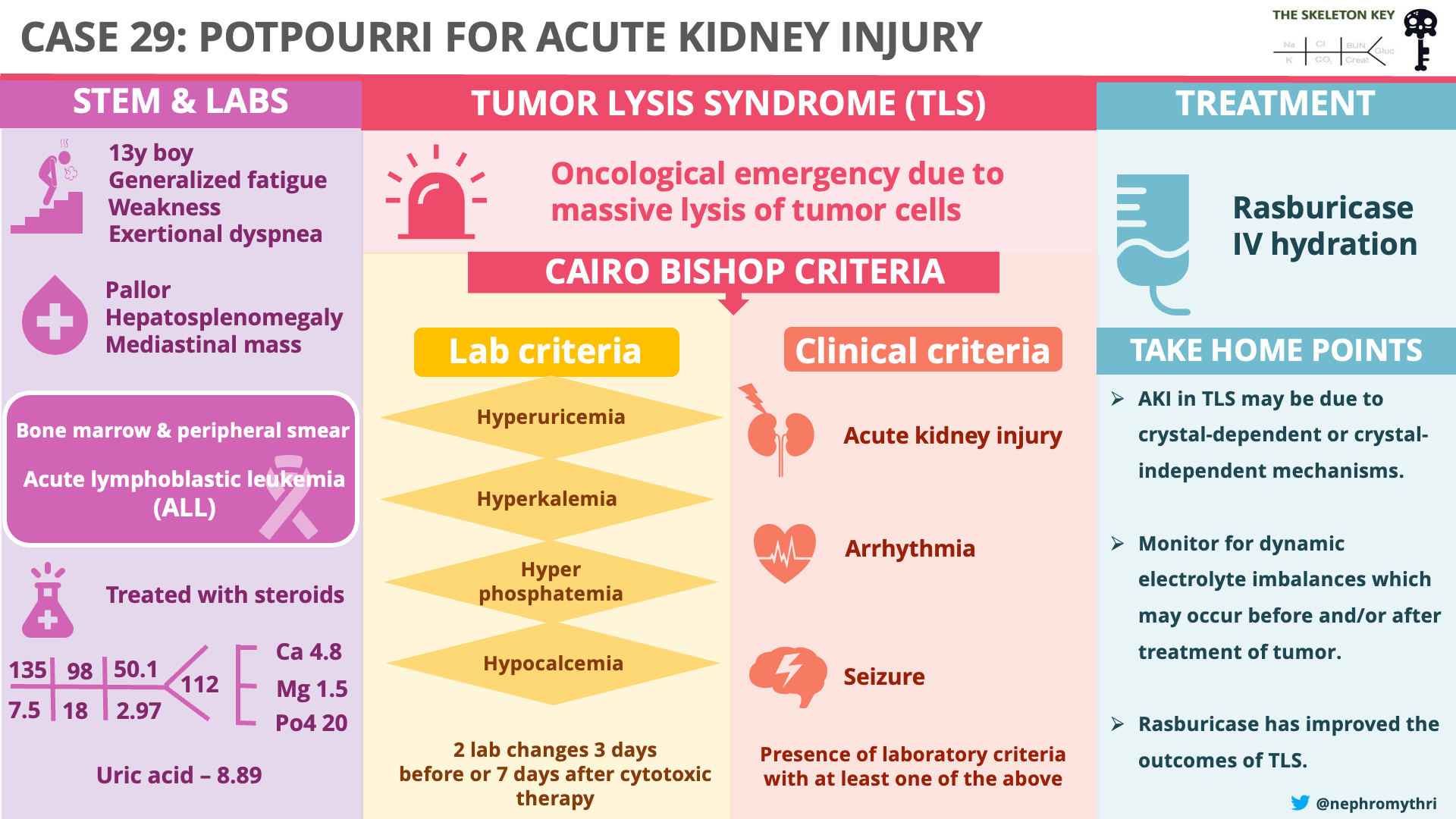
A 13-year-old boy with no past medical history presented to the oncology services with one-month of intermittent fever and exertional dyspnea associated with anorexia, fatigue, and weakness. He had weight loss of over 10% in 6 months.
Vital signs included HR 96 bpm, BP 110/70 mm Hg. On physical examination, the child looked pale and volume depleted. He had a normal cardiopulmonary examination and hepatosplenomegaly.
B. The Labs
Blood reports showed a hemoglobin of 6.3 g/dL, a total WBC count of 210,000/ mm3, and a platelet count of 10,000/mm3. Chest X-ray revealed a mediastinal mass. Peripheral smear and bone marrow examination were suggestive of B-cell type acute lymphoblastic leukemia (ALL).

The metabolic profile demonstrated hypercalcemia, hypophosphatemia, hyperuricemia, and acute kidney injury (AKI) with high anion-gap metabolic acidosis.
C. The Workup
ALL is a hematologic malignancy characterized by excessive production of immature leukocytes by the bone marrow and is the most common malignancy in children younger than 15 years.
Why the low phosphate?
This patient had a phosphate level of 1.0 mg/dL (normal range 2.4‐5.1 mg/dL).The approach to hypophosphatemia can be reviewed in Skeleton Key Group Case #25.
But what is the association between ALL and phosphate? Hypophosphatemia can occur in acute leukemia due to phosphate uptake by the rapidly dividing leukemic cells, though it was likely multi-factorial in this case.
Why the elevated calcium?
This patient had a total serum calcium level of 12.7 mg/dl ( normal range 8.5-10.5 mg/dl). His parathyroid hormone (PTH) level was low, suggesting a PTH-independent process for hypercalcemia. Vitamin D levels were within normal limits. We inferred that this patient might have had malignancy-associated hypercalcemia. This is usually caused by three main mechanisms – excessive secretion of PTHrP, release of osteoclast activating factors by bony metastasis and by excessive production of calcitriol. A more in-depth review of calcium balance is discussed previously in Skeleton Key Group Case #10.
How about the uric acid?
This patient had a uric acid of 28 mg/dL (normal range 3.5-7 mg/dL). Intracellular purine nucleic acids are catabolized to xanthine and hypoxanthine, which get converted to uric acid by the enzyme xanthine oxidase. Hyperuricemia may be related to increased production or decreased excretion of uric acid, increased purine intake, or rarely hereditary conditions associated with altered purine nucleotide synthesis. In this case, it was likely due to a degree of spontaneous leukemic cell lysis and overproduction of uric acid.
And finally, acute kidney injury!
This patient had a creatinine of 3.4 mg/dl (normal range 0.4 -1.3 mg/dl). The cause of acute kidney injury is likely multifactorial, with causes including hypovolemia and uric acid precipitation.
Uric acid is poorly soluble in water and precipitates in an acidic environment (like distal tubules and collecting ducts). Whenever there is overproduction of uric acid, such as in leukemias, it can precipitate in the kidney tubules causing obstruction and acute kidney injury, which causes a vicious cycle as GFR drops and the body is less able to excrete uric acid. Uric acid also directly exacerbates acute kidney injury by causing vasoconstriction and impairing autoregulation, reducing blood flow to the kidneys. This spontaneous release of uric acid can then worsen when chemotherapy is provided and more cancer cells undergo lysis
D. More Data
The patient was started on steroids for ALL and rasburicase in the context of existing hyperuricemia and being high risk for development of tumor lysis syndrome (TLS). Following this, he developed persistent hyperphosphatemia and hyperkalemia (see table below), and a nephrology consult was sought for consideration of kidney replacement therapy. The labs at this point were as follows:
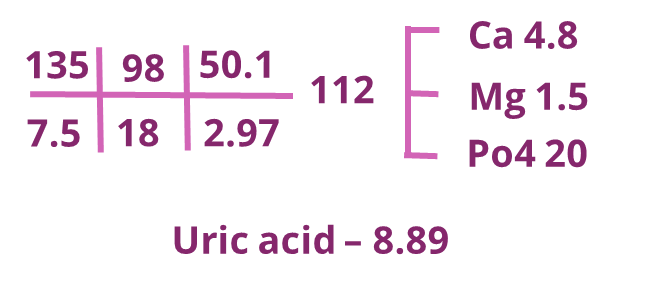
Pseudohyperkalaemia was considered in the differential, as it is well-recognised that fragile leukocytes can be broken down due to mechanical stress and release intracellular potassium, especially if the time to lab processing is prolonged. This was ruled out using immediate processing of a venous blood gas sample, confirming true hyperkalemia.

Table showing everyday representative lab values. (Note: lab values were repeated more often for monitoring)
E. The Final Diagnosis
Tumor lysis syndrome (TLS) is an oncological emergency due to the massive lysis of tumor cells, releasing vast amounts of potassium, phosphorus, and nucleic acids into the blood. Note in our patient the very large, rapid rise in phosphate and a corresponding drop in calcium, along with their spike in potassium.
Tumors with high risk for TLS have high tumor burden, proliferative rate, and chemosensitivity. TLS is usually associated with hematologic malignancies such as non-Hodgkin’s lymphoma (NHL), acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia (CLL), and germ cell tumors. Solid tumors and slow-growing hematologic malignancies such as multiple myeloma (MM) have a much lower risk for TLS.
TLS can occur either spontaneously or after the initiation of cytotoxic therapy. It usually occurs following treatment with steroids, chemotherapy, cytolytic antibodies, or radiation therapy due to rapid breakdown of tumor cells. AKI in TLS can be caused by crystal-dependent and crystal-independent mechanisms. First, let’s look into crystal dependent mechanisms. Nucleic acids get broken down to uric acid, which may precipitate in renal tubules causing acute tubular injury. Increased phosphate levels combine with calcium and deposit calcium phosphate in the kidney’s tubules, also causing acute kidney injury. Uric acid precipitates readily in the setting of calcium phosphate crystals and calcium phosphate precipitates readily in the setting of uric acid crystals, thus causing AKI. This phenomenon is known as epitaxy, in which there is a growth of one type of crystal over another. Calcium phosphate coats the otherwise soluble uric acid. These types of cystals usually contain a mixture of minerals. Crystal independent mechanisms of AKI are due to histones and cytokines. Histones are found in the nucleus of eukaryotic cells. They are basic proteins that combine with DNA and help condense it to chromatin. During TLS, extensive quantities of extracellular histones are released into the circulation. These histones are cytotoxic and cause endothelial injury leading to AKI in TLS. This phenomenon has been demonstrated in a mice model by Marine Arnaud et al.
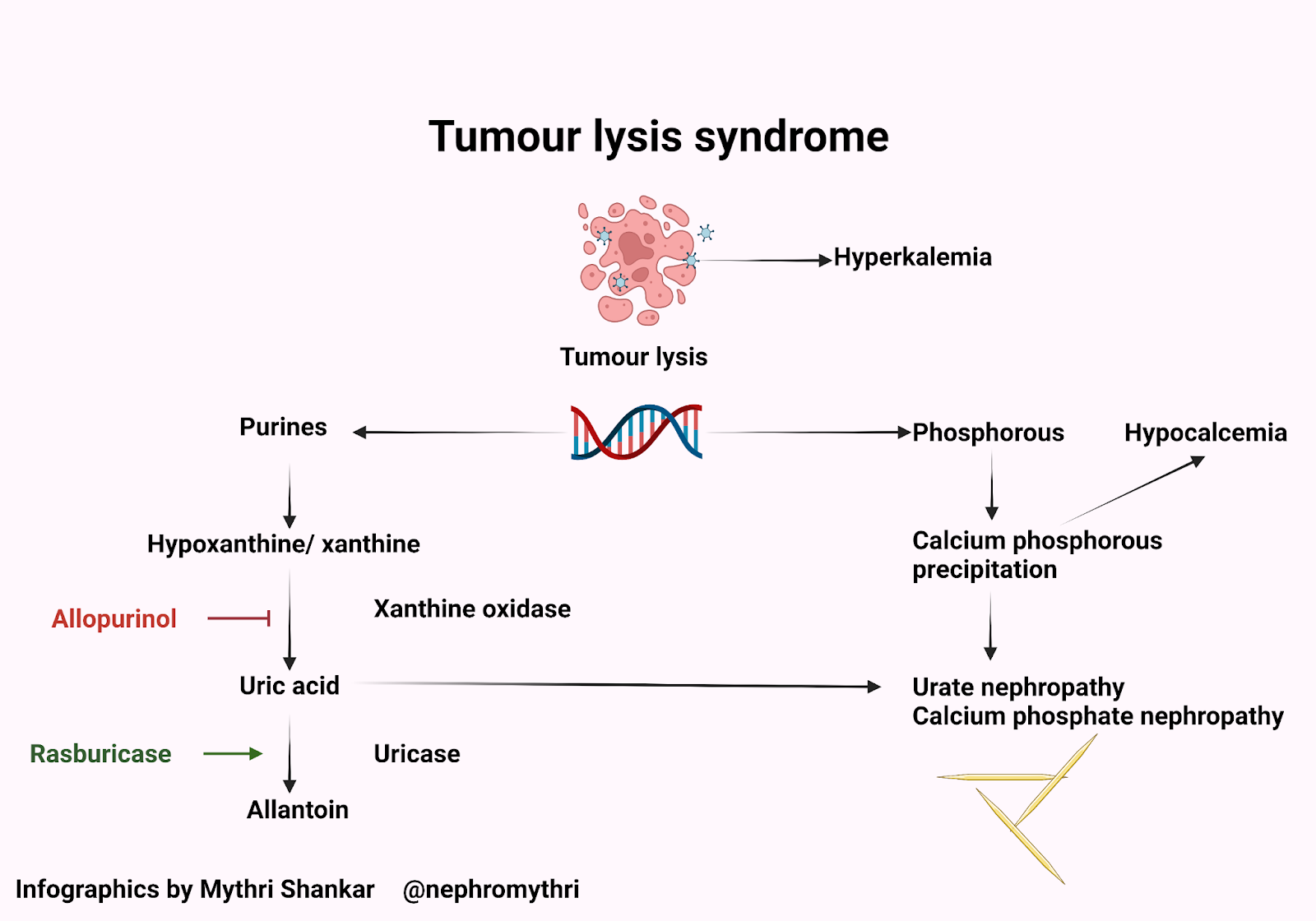
Diagnostic criteria: Cairo-Bishop definition (2004)
TLS is traditionally categorized as clinical TLS and laboratory TLS using the Cairo-Bishop definition.
| Variable | Value | Change from baseline value |
| Uric acid | ≥8 mg/dL (≥476 mmol/L) | 25% increase from baseline |
| Potassium | ≥6.0 mEq/L (≥6 mmol/L) | 25% increase from baseline |
| Phosphorous | ≥4.5 mg/dL (≥1.45 mmol/L) for adults≥6.5 mg/dL (≥2.1 mmol/L) for children | 25% increase from baseline |
| Calcium | ≤7 mg/dl (≤1·75 mmol/L) | 25% decrease from baseline |
Adapted table from Cairo et al.
Clinical TLS: Presence of laboratory criteria with at least one of the following: acute kidney injury, cardiac arrhythmia, or seizure.
These criteria are broadly used, though there are several drawbacks, especially from a nephrology point of view. First, these criteria are defined relative to the timing of cytotoxic therapy. Hence, it does not consider spontaneous tumor lysis syndrome. Second, it defines AKI as 1.5 times the upper limit of serum creatinine (adjusted for age and sex), which is a poor indicator of true AKI and can include patients with chronic kidney disease as well. Third, the distinction between laboratory criteria and clinical criteria is of limited clinical significance as most patients are initiated on treatment as soon as the lab abnormalities develop. Fourthly, some feel they emphasize a biochemical definition that is non-specific ( for example, high potassium and phosphate being extremely common with any cause of AKI) whereas it is the history that most informs the diagnosis.
In this case, the patient had AKI prior to initiation of chemotherapy, but his labs are showing hypophosphatemia and normokalemia, suggesting against gross spontaneous TLS. Plus, he had alternative reasons for AKI (e.g. volume depletion, hypercalcemia). However, after starting steroid therapy he developed rapid and severe electrolyte disturbances, likely exacerbated by the pre-existing low glomerular filtration rate, in keeping with chemotherapy-induced TLS.
Lysis of tumor cells leads to hyperphosphatemia. The phosphate content of malignant cells is four times more than the other cells. Hyperphosphatemia is less common in spontaneous TLS because rapidly growing tumor cells take up phosphate that is released from the dying tumor cells, causing net phosphate flux to be neutral. The rapid rise in serum phosphate after chemotherapy causes secondary hypocalcemia as calcium and phosphate bind in the circulation and deposit in tissues. Severe secondary hypocalcemia can cause tetany, seizures, and arrhythmias. When the Ca X PO4 product > 60mg2/dL2, there is an increased risk of precipitation of calcium phosphate crystals in the renal tubules causing AKI. Some see Ca X PO4 >70 mg2/dL2 as an indication for kidney replacement therapy (KRT). Generally, we tend to tolerate asymptomatic hypocalcemia without replacement due to the risk of worsening calcium phosphate precipitation. Hence we use non-calcium-containing phosphate binders, such as sevelamer, which may be useful in reducing phosphorus levels in TLS. Sevelamer reduces phosphate absorption from the digestive tract. Few retrospective studies have found it useful in the treatment of hyperphosphatemia secondary to TLS. However, randomized control trials are required to confirm its role in reducing the need for KRT and mortality in patients with TLS.
F. Management
Risk stratification of malignancy is essential to guide therapy. We have to take prophylactic measures in patients at high risk for TLS such as those with dehydration, hyperuricemia, and pre-existing kidney disease. The primary prophylactic measures are IV hydration and hypouricemic agents (rasburicase). Since TLS is a very dynamic process, the importance of monitoring the lab parameters cannot be over-emphasized.
IV hydration aims to increase the urine output to prevent the precipitation of uric acid and calcium phosphate crystals in the tubules.
Choice of fluid: Normal saline is largely considered in patients who are volume-depleted. As these patients are at risk of precipitation of calcium phosphate stones, any fluid with calcium should be avoided. Urine output should be closely monitored and (if possible) maintained at 2 ml/kg/hour or atleast 3 litres per day. IV hydration therapy should be continued until the tumor burden is largely resolved, there is no ongoing TLS, and patients can drink adequate fluids with good urine output.
Uric Acid Lowering
Rasburicase is a recombinant urate oxidase. It breaks down uric acid into allantoin, which is water-soluble and non-nephrotoxic. On the other hand, allopurinol, a xanthine oxidase inhibitor, causes xanthine accumulation and xanthine crystal formation. Rasburicase is currently the preferred drug for hyperuricemia in TLS. Rasburicase is contraindicated in patients with G6PD deficiency. This is because in patients with G6PD deficiency, uric acid metabolism leads to hydrogen peroxide generation leading to methemoglobinemia and, in severe cases, hemolytic anemia. Hemolysis usually occurs 2 to 3 days after starting rasburicase. Monitor the patient closely for hemolysis and discontinue rasburicase as soon as hemolysis is detected. Many oncologists test for G6PD deficiency before giving chemotherapy in malignancies at high risk of TLS, especially in those of Mediterranean or African ancestry. Though it is highly potent in reducing uric acid levels, its availability is limited in low-resource settings. If only humans had retained their urate oxidase gene, we wouldn’t have uric acid nephropathy and gout!
Also, it is important to note that the post-rasburicase uric acid level for monitoring needs to be processed urgently (ideally in a lithium heparin tube, and on ice) to stop the rasburicase from continuing to chew up the uric acid and give a falsely low result.
Urine alkalinization
This approach is no longer favored as a means to prevent uric acid nephropathy, especially as many countries have access to rasburicase, meaning that addressing the risk of calcium phosphate crystals often takes precedence over uric acid crystal prevention. One experimental study showed that maintaining high distal urine flow with either water or solute diuresis was as effective as alkalinization of urine in preventing uric acid precipitation and stones. Meanwhile, alkalinization therapy promotes the precipitation of calcium phosphate crystals in the kidneys, heart, and other organs, which can be detrimental. Also, alkalinization can exacerbate hypocalcemia, increasing the risk of seizures, tetany, and arrhythmia.
Diuretics
Enhanced urine flow is postulated to reduce the precipitation of calcium phosphate and uric acid crystals. Using diuretics to enhance urine flow in TLS has not been studied rigorously. Thus, diuretics are reserved only in the setting of volume overload and are not used prophylactically.
Kidney replacement therapy (KRT)
Dialysis may be required in severe AKI with life-threatening hyperkalemia, hyperphosphatemia, and hyperuricemia. If there is ongoing liberation of these electrolytes in actively lysing cells, continuous kidney replacement therapy (CKRT) is often preferred over conventional intermittent dialysis as it is better able to avoid “rebound” hyperkalemia and hyperphosphatemia that can occur between intermittent hemodialysis treatments. However, minute-to-minute clearance of potassium is better with conventional intermittent dialysis, so for life-threatening hyperkalemia, the best option is to use intermittent hemodialysis to rapidly lower potassium to safe levels, and then continue with CKRT to avoid rebound hyperkalemia from ongoing cell lysis.
In this patient, medical management was initiated as there were no life-threatening indications and the patient was non-oligoanuric. Also, he had low platelet counts which was a contraindication for hemodialysis catheter insertion. Hence, he was closely monitored for biochemical and cardiac parameters in the intensive care unit, with a low threshold to initiate intermittent dialysis if there was no rapid response to medical management. He was initiated on hydration therapy with IV isotonic saline which was continued for 2 days until recovery of kidney function. Urine output was maintained at 2 mL/kg/hr during the treatment. Subsequently, kidney function normalized in 3 days.

G. Take Home Message
TLS is an oncological emergency arising from the successful treatment of malignancy. The pathogenesis of AKI in TLS may be due to crystal-dependent or crystal-independent mechanisms. Dynamic electrolyte imbalances and acute kidney injury may occur before and after treatment, keeping the nephrologists on their toes. Rasburicase has improved the outcomes of TLS, and the use of KRT has its place in managing life-threatening electrolyte imbalances.
Reviewed by Joel Topf, Chi Chu, Dominique Tomacruz, Dilushi Wijayaratne, Raad Chaudary, Jefferson Triozzi, Matt Sparks

Nice explanations of the various concepts! Great review
Very well!
Really interesting case