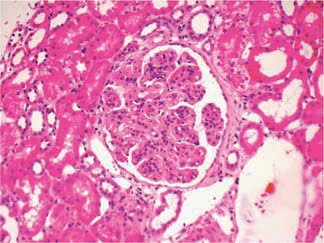
We recently saw a case of adult-onset dense deposit disease (DDD) at pathology conference. DDD is rare, affecting 1-3 per million people. For historical reasons it has been classified under the membranoproliferative subgroup of disease patterns (as type 2), but many voices within pathology are calling for this to be changed.
DDD primarily presents in childhood with the nephrotic or nephritic syndrome. However, it can have a wide range of other clinical presentations, including sterile pyuria, isolated macroscopic hematuria, microscopic hematuria with subnephrotic range proteinuria, and isolated proteinuria. Most patients have low C3 levels; C4 is usually normal and around 80% have a detectable C3 nephritic factor. As a rule of thumb, C3 is nearly always depressed in children, whereas this occurs less than half the time in adult presentations.
Extra-renal manifestations include formation of retinal drusen and development of acquired partial lipodystrophy. Lipodystrophy is highly correlated with the presence C3 nephritic factor, with detectable levels in almost all cases.
Extra-renal manifestations include formation of retinal drusen and development of acquired partial lipodystrophy. Lipodystrophy is highly correlated with the presence C3 nephritic factor, with detectable levels in almost all cases.
DDD is thought to be caused by excess activation of the alternative complement pathway. In the alternative pathway, the normal cascade is initiated by cleavage of C3 to C3a and C3b. C3b joins with Factor B and properdin to form C3 convertase. C3 convertase causes further cleavage of C3, increasing the amount of C3b available to form C5 convertase. C5 convertase cleaves C5 to C5a and C5b. The former has a chemoattractant function; the latter contributes to formation of the membrane attack complex. Factor H serves to regulate this pathway by promoting degradation of C3 convertase and C5 convertase. It also combines with Factor I to inhibit C3b.

Thus, it is thought DDD can arise from two main events: formation of a C3 convertase stabiliser (C3 nephritic factor) or loss of Factor H (mutation v acquired). The formation of excess complement components allows deposition in the GBM and incitement of an inflammatory reaction.
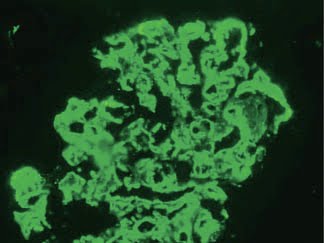
The diagnosis is made by examination of renal histology. Interestingly, only 25-44% of cases present with a membranoproliferative pattern of injury. Immunofluorescence is generally positive for C3 in areas of deposits. Immunoglobulin staining is typically negative, which helps distinguish it from MPGN type 1. The pathognomonic changes are seen in electron microscopy – abnormal electron-dense deposits in the GBM.
Finally, a first presentation of DDD in an adult should always prompt a search for an underlying plasma cell dyscrasia, which was discovered in this case.
The diagnosis is made by examination of renal histology. Interestingly, only 25-44% of cases present with a membranoproliferative pattern of injury. Immunofluorescence is generally positive for C3 in areas of deposits. Immunoglobulin staining is typically negative, which helps distinguish it from MPGN type 1. The pathognomonic changes are seen in electron microscopy – abnormal electron-dense deposits in the GBM.
Finally, a first presentation of DDD in an adult should always prompt a search for an underlying plasma cell dyscrasia, which was discovered in this case.
A review of the complement cascade by Nate can be found here.
Finnian McCausland M.D.
