Summer has come to the northern hemisphere and it is time to praise our kidneys for all the hard work of preserving the intravascular volume. Although only 30% of Na reabsorption takes place in the distal nephron, this is the place for most common genetic diseases causing abnormal sodium handling:
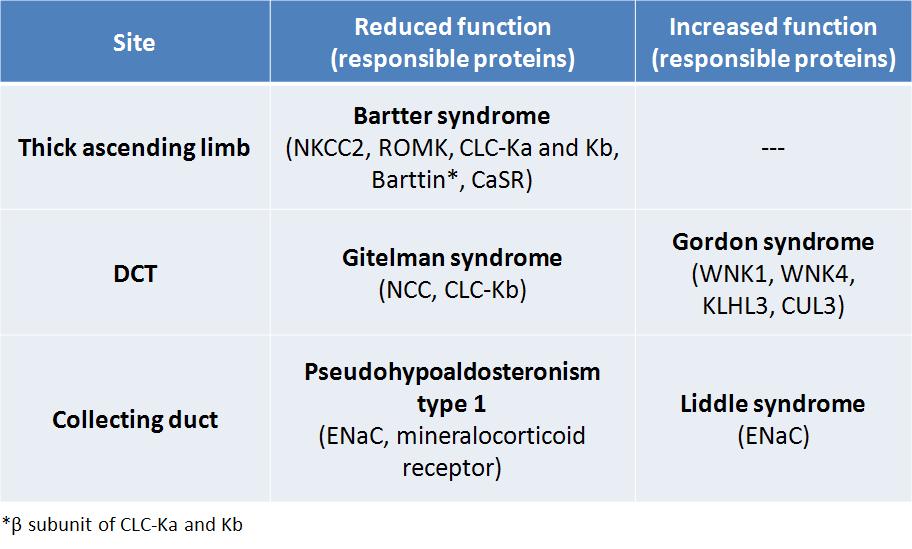
What are the responsible proteins for these diseases?
1. Bartter syndrome: Na reabsorption decreases via NKCC2 due to defects in NKCC2, ROMK and basolateral Cl channels or from an overactive basolateral CaSR.
2. Gitelman syndrome: Na reabsorption decreases due to a reduced number of NCCs on the luminal membrane.
3. Pseudohypoaldosteronism type 1: Na reabsorption decreases due to the defects in ENaC or mineralocorticoid receptor.
4. Liddle syndrome: Na reabsorption increases due to a higher number of ENaCs on the luminal membrane (they are spared from ubiquitination).
5. Gordon syndrome: Na reabsorption increases due to a higher concentration of NCCs on the luminal membrane (defects in WNK4 or overactive WNK1). More recently, mutations were discovered in the genes encoding KLHL3 and CUL3 proteins.
CUL3 is a component of ubiquitin ligase and KLHL3 is its partner (expressed in the DCT). It is speculated that the defects in these proteins may change the distribution of NCC.
Is this clinically relevant?
In Gordon syndrome, these mutations can occur de novo and are encountered more frequently (47% for KLHL3 and 32% for CUL3) than those involving the WNK kinases (13%). It appears that they are under-diagnosed and some of the cases of RTA type 4 with hypertension could be caused by them!
50 years after Bartter syndrome was described, the work on mechanisms responsible for abnormal Na regulation in the nephron continues…
Posted by Tomoki Tsukahara MD


This is named after a nephrologist at Yale who is still on faculty at the VA in west haven.
I would add Gain-of-function mutation of the mineralocorticoid receptor (AKA "Geller Syndrome") to increased function in collecting duct. This condition usually causes exacerbation of hypertension during 2nd or 3rd trimester of pregnancy when progesterone levels are high. Spironolactone is innefective and can exacerbate hypertension in this condition.