
Angiotensin II inhibits the Na-K-ATPase leading to cell depolarization, calcium influx into cells and aldosterone secretion. Similar effects are seen when cells are treated with oubain, a specific Na-K-ATPase inhibitor that also leads to hyperaldosteronism.
In 2011 in a seminal paper in Science, Choi et al reported finding somatic mutations in KCNJ5, a membrane potassium channel in patients with APA. These were identified by sequencing tissue from the tumors and comparing with the surrounding tissue. Subsequently, it has been found that about 30-40% of patients with APA have somatic mutations in KCNJ5. These mutations are believed to reduce the ion selectivity of the channels, allowing Na to move into the cell and reduce the resting membrane potential. A number of families have been identified with KCNJ5 mutations resulting in bilateral hyperplasia – now called Familial Hyperaldosteronism type III.
Recently, a paper was published in Nature Genetics which attempted to determine if there were other somatic mutations in patients with APA. In this study, they took KCNJ5-normal patients and sequenced the exons of the tumors and the surrounding tissue. There were very few mutations identified but 5/9 patients had mutations in ATP1A1, a component of the Na-K-ATPase or ATP2B3, a component of the calcium ATPase that removes calcium from adrenal cells. Follow-up targeted sequencing of 300 patients with APA revealed that about 7% had mutations in one of these two genes. Patients with these mutations had higher aldosterone levels, lower minimum potassium levels and higher systolic BP, all indicators of more severe disease. Notably, no families have been identified with these mutations. In vitro studies revealed that cells with these mutations have very low membrane potentials and it is speculated that if this was a germline mutation, it would likely not be compatible with life. This is a fascinating insight into how very small changes in electrolyte channels can have far-reaching consequences and shows a great progression from exome sequencing to the bench and to clinical investigation.
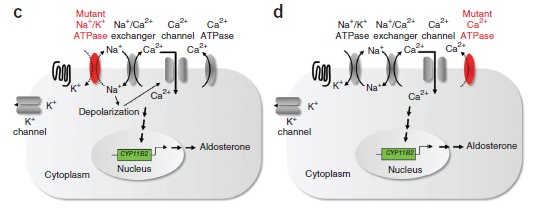
The images in this post are taken from the recent paper in Nature Genetics. One would wonder if somatic mutations explain some of the missing heritability that were are seeing in genetic studies of common diseases. See this previous post by Lisa on the genetic causes of hypertension.

