Hypertension is commonly associated with chronic kidney disease (CKD). The combination of hypertension and CKD together make for a BIG clinical problem. The last two decades have seen research focused on many potential causative pathways linking both hypertension and CKD. In this blog post, we will review advances in our understanding of how kidney inflammation might play a role in hypertension.
The proposed role of inflammation in hypertensive kidney disease has strong theoretical support and is not a new idea. Olsen described an inflammatory mononuclear cellular infiltration in arterioles and arteries of patients with hypertension. The author observed the presence of inflammatory cells in vessels in differing stages of degeneration. Mononuclear cells include monocytes and lymphocytes but exclude neutrophils and other granulocytes. While scientists had suggested inflammation as a culprit in hypertensive vascular disease as early as 1972, many felt that hard evidence for the argument that intrarenal inflammation may cause or promote high blood pressure was lacking, until recently (Norlander et al. J Exp Med 2018; Itani et al. Hypertension 2016; Loperena et al. Cardiovasc Res 2018). There are three central arguments supporting a role for kidney inflammation in hypertension.
First, inflammation is everywhere, and that is not necessarily a bad thing. We often think of inflammation as something that causes pain, for example the pain experienced after an ankle sprain while playing sports is caused by inflammation. Inflammation is something we can take a Tylenol or Advil to reduce the pain and allow us to get along with our day. That is definitely true, but let’s dive in a little deeper. Inflammation is a natural biological process that, in addition to fighting infectious pathogens and foreign particles, allows our bodies to respond to cell injury and death. It is a healing process as much as it is one that damages tissue and causes pain. One comparison is that inflammation is like the yin and yang from Chinese philosophy. There is a dark side of inflammation that causes tissue damage, and there is a light side that is necessary and responsible for healing. An appropriate balance must be struck in order to execute optimal healing. In hypertensive kidney disease, there is an increase in cell injury, cell death, and release of inflammatory signaling molecules called cytokines.
Therefore, it logically follows that inflammation would be involved in the processes of clearing dead cells, providing growth factors for new cells, and in responding to cytokines, all of which occur in hypertension-associated kidney disease. In response to these signals, inflammation can cause tissue damage and blood vessel fibrosis. It remains unclear whether the earliest inciting events in hypertensive kidney disease damage are caused by inflammatory cells, hydrostatic pressure changes, in response to circulating signaling molecules, or changes in nervous system inputs which partially control the diameter of blood vessels. It is likely that multiple pathophysiologic mechanisms are active simultaneously.
Second, when researchers study the kidney tissue of individuals that have experienced hypertensive kidney disease, they can see inflammatory cells. Inflammatory cells are simply white blood cells that carry out much of the process of inflammation. When we study hypertension-associated kidney disease, a mixture of cell types can be observed histologically. However, two cell types appear to be functionally predominant early in hypertension-associated kidney disease, antigen presenting cells and T cells (Norlander et al. J Exp Med 2018; Itani et al. Hypertension 2016; Loperena et al. Cardiovasc Res 2018). One immediate question is whether the inflammatory cells are causing hypertensive kidney disease, promoting it in some way, or are the simply bystanders, called to action because of cell injury or death that may have already occurred?
Drs. Guzik, Harrison, and colleagues demonstrated that T cells play a causative role in angiotensin II-induced hypertension and vascular dysfunction. The authors utilized a genetic mouse model wherein the recombination activating gene 1 (RAG1) is deleted, and the animals lack T cells and B cells (two types of inflammatory cells). The authors treated the RAG1 deficient mice with angiotensin II, a molecule which normally increases blood pressure. When T cells and B cells were not present, the mice did not demonstrate hypertension. In order to determine whether T cells or B cells were responsible for angiotensin II-induced hypertension, they added each cell type back and found that T cells were responsible. If the T cells lacked the receptor for angiotensin II, the transferred T cells did not restore the hypertension. Therefore, T cells, and their response to angiotensin II, were responsible for causing hypertension and vascular damage and dysfunction, at least in the case of this animal model of hypertension. While angiotensin II-induced hypertension is a good disease model, it does not capture the complexity of hypertension in human patients.
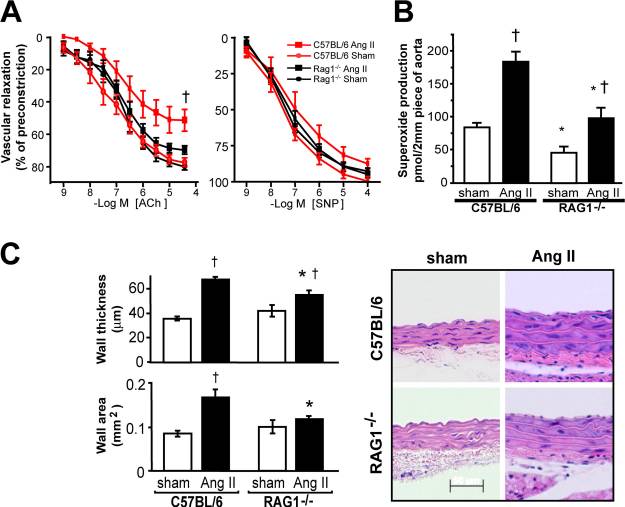
Drs. Crowley, Ruiz, and colleagues used another type of immune deficient mouse called severe combined immunodeficiency (SCID) mice in angiotensin II-induced hypertension. SCID mice, also deficient in T and B cells, were also protected from hypertension and associated heart and kidney disease in the setting of long-term treatment with angiotensin II. This study further supported a role for kidney inflammation in causing hypertension and therefore in causing hypertension-associated kidney disease.
Interestingly, a recent study in Hypertension in 2017 reported a loss of the protection from hypertension in the setting of T cell deficiency. The authors suggest that this may be due to a change in the genetics of the RAG1-deficient inbred mouse model wherein the angiotensin receptor expressed by kidney cells was increased, canceling out the protective effect of T cell deficiency. Therefore, the conclusion that T cells are necessary for the genesis of angiotensin II-induced hypertension remains valid. However, this example highlights the importance for scientific rigor and reproducibility.
Antigen presenting cells and T cells can work together in generating a chronic inflammatory response. Dr. Harrison and his research group found that isolevuglandin-modified proteins were perceived by the immune system as foreign antigens and were important in driving hypertension-associated kidney inflammation and damage. This represents an important advancement in our understanding of the cellular and molecular mechanisms of inflammation and hypertensive kidney disease. The T cell and antigen presenting cell activation due to these modified proteins may be targeted by experimental therapies in the future.
Third, signaling molecules, called cytokines, are made by inflammatory cells and kidney cells in patients with hypertension. Cytokines have a diverse array of functions from promoting inflammatory damage to promoting healing. Cytokines can also affect transporter proteins which are essential for kidney regulation of plasma electrolytes. Scientists have found that, in the setting of hypertension, activated inflammatory cells release cytokines both locally in the kidney and into the circulation (Norlander et al. J Exp Med 2018; Dorffel et al. Hypertension 1999; Bautista et al. J Hum Hypertens 2005; Dalekos et al. J Lab Clin Med 1997). Interestingly, not all patients with hypertension present with increases in circulating pro-inflammatory cytokines, which represents a gap in our knowledge of the relationship between inflammation and hypertensive kidney disease.
Future studies will aim to take advantage of our new knowledge of how kidney inflammation and hypertension are related. It is possible that the disease can be fought by patients and their doctors using new targeted medications, such as with biologics (drugs using proteins or live cells) or nanomedicine (drugs targeted at specific cell types) approaches, in addition to diet and exercise changes. Previous clinical trials testing the effects of non-steroidal anti-inflammatory drugs (NSAIDs) on blood pressure control in patients with hypertension demonstrated either increases or no change in measured blood pressure (Whelton et al. Am J Cardiol 2002; Sowers et al. Arch Intern Med 2005; Houston et al. Arch Intern Med 1995). This suggests that NSAIDs, although anti-inflammatory, are not efficacious for improving high blood pressure. This may be due to the fact that a primary action of NSAIDs on blood pressure is via inhibition of prostaglandin synthesis. Prostaglandins, in turn, signal to kidney cells and can generally be categorized as antihypertensive.
The NSAID example illustrates that new creative approaches will be necessary to intervene in inflammation in hypertensive kidney disease. Further, one of the main goals in inhibiting intrarenal inflammation in hypertension will be to protect the kidney tissue from inflammatory damage. Therefore, an important outcome of future studies would be the long-term preservation of kidney function.
One thing is clear, kidney inflammation does play a role in hypertension. Because the work regarding T cells and antigen presenting cells is in early stages of translation to human biology (can be considered “basic science” or “pre-clinical”), it is not clear how these results will be translated to the clinic. However, the future is bright. As a result of this exciting work, we have a new appreciation for the relationship of the immune system, hypertension, and the kidney. The tale of two cell types, T cells and antigen presenting cells, is henceforward inextricably tied to hypertension and kidney inflammation.
Jeremie Lever
MD/PhD Candidate
University of Alabama at Birmingham

Thank you for the feedback, Dr. Steiner!
Jeremie
A really nice review and persuasive argument. But as for the overall contribution to loss of GFR, in prospective studies in well screened populations, true hypertensive ESRD is vanishingly rare (Hsu Curr Opin Neph Hyperten 2011) . Hoerger (AJKD 2010) could predict all the yearly ESRD in the US by assuming that the incremental loss of GFR in hypertensive populations was less than 1 cc/min/decade. Primary renal disease causing hypertension is another matter. This dominates retrospective databases and creates a different impression (Schlessinger AJKD 1994). These are only a few of the references that could have been cited here.
Bob Steiner