Acute interstitial nephritis (AIN) is a common cause of acute kidney injury (AKI), estimated to account for 15-20% of cases of AKI. The term AIN does not refer to a specific disease, but rather indicates an immune-mediated form of tubulointerstitial injury which may result from a variety of underlying causes. Drug hypersensitivity accounts for the majority of cases (~70%), with the remaining cases attributable to systemic or direct renal infections, autoimmune diseases, hereditary and metabolic disorders, obstruction/reflux, or unknown causes. AIN also frequently accompanies glomerulonephritis and/or vasculitis. Notably, hematologic malignancies and lymphoproliferative disorders can directly infiltrate the renal parenchyma and closely mimic AIN.
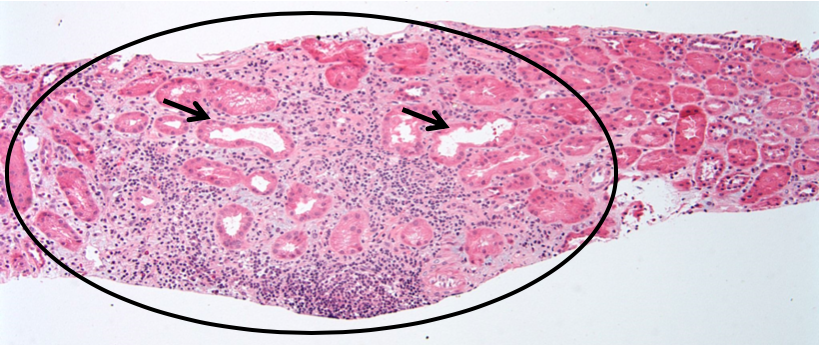
Clinically, AIN typically manifests as acute kidney injury and may be accompanied by other systemic symptoms depending on the underlying cause. The main histologic finding in AIN is a mixed interstitial inflammatory infiltrate, often associated with tubulitis and interstitial edema. The appearances are often similar despite varied mechanisms, but there are certain pathologic clues which may suggest a particular etiology, detailed below.
Drugs
The incidence of drug-induced AIN appears to be increasing, with over 150 different drugs identified as culprits. Drug-induced AIN typically represents an idiosyncratic hypersensitivity reaction which is not dose dependence, and is often associated with systemic hypersensitivity manifestations such as rash, fever, and eosinophilia. Other mechanisms have also been implicated, such as altered T cell regulation in checkpoint inhibitor therapy. The most common offending agents include antibiotics, proton pump inhibitors, nonsteroidal antiinflammatory drugs, salicylates, and chemotherapeutic agents.
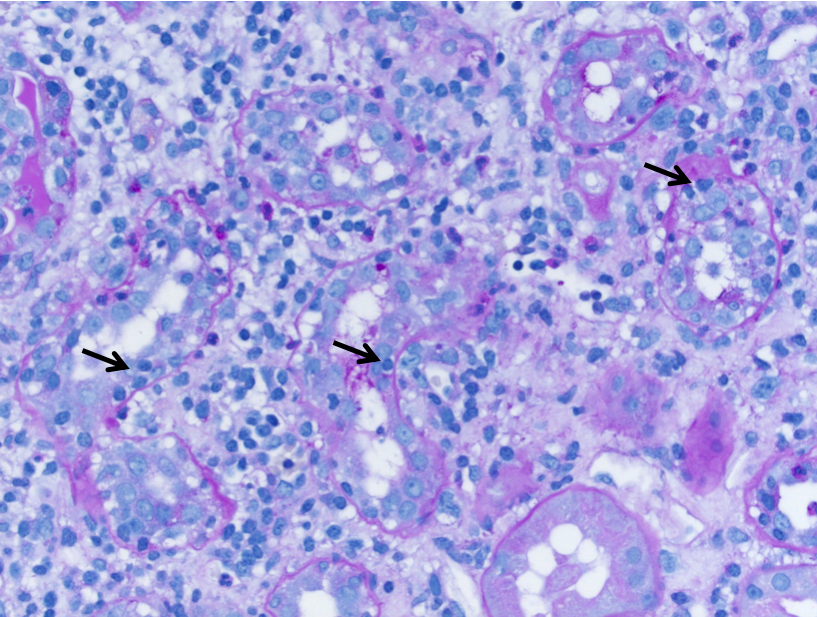
By light microscopy, drug-induced AIN is characterized by mononuclear cell inflammation involving the interstitium and tubules (“tubulitis”) composed mostly of T lymphocytes. There are also scattered plasma cells and macrophages, and variable numbers of eosinophils. Frequently there is patchy interstitial edema, and concomitant features of acute tubular injury. There may be interstitial granulomas with epithelioid histiocytes and rarely multinucleated giant cells; in fact, drug hypersensitivity is the most common cause of granulomatous interstitial nephritis. Granulomatous interstitial nephritis can also be seen in sarcoidosis and certain mycobacterial and fungal infections. Clues to a drug hypersensitivity reaction as the underlying etiology of AIN include a prominent eosinophilic infiltrate, interstitial inflammation which is more prominent in the medulla (where the drug is more concentrated), and the temporal relationship between the development of acute renal insufficiency and exposure to a new medication. Other possible lesions of drug-induced kidney injury which may be seen in conjunction with AIN include papillary necrosis, minimal change disease, and membranous nephropathy.
Infection
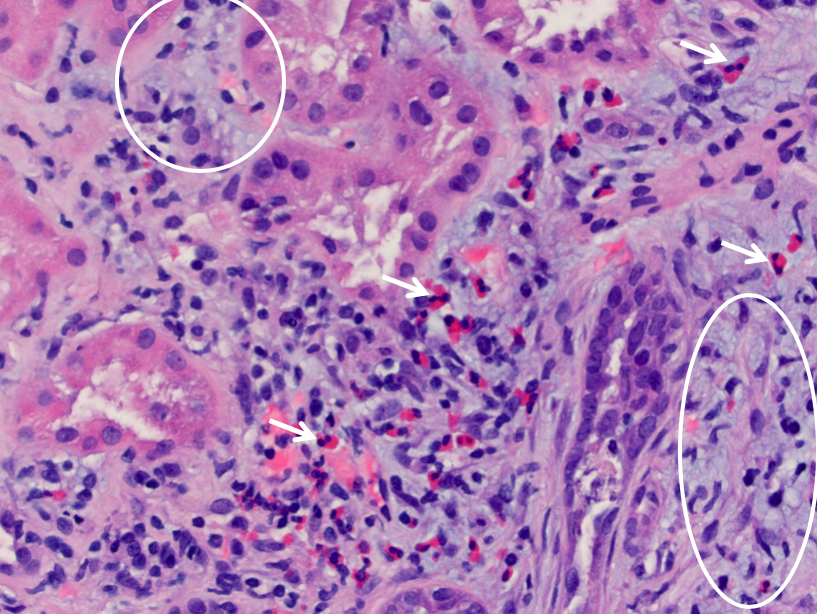
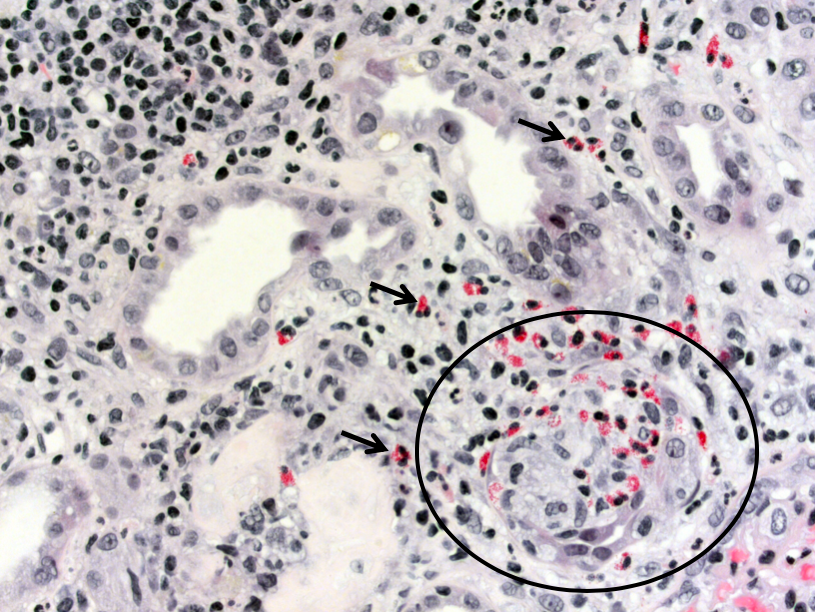
Acute pyelonephritis is usually an ascending infection caused by Gram negative bacteria from the gastrointestinal tract, particularly Escherichia coli. Presentation typically involves fever and flank pain, as well as evidence of acute kidney injury. Recurrent episodes can result in chronic pyelonephritis with progressive scarring and renal insufficiency. By light microscopy, acute pyelonephritis is characterized by prominent aggregates of neutrophils and cellular debris within the tubular lumens (“pus casts”), as well as neutrophilic interstitial inflammation and tubulitis.
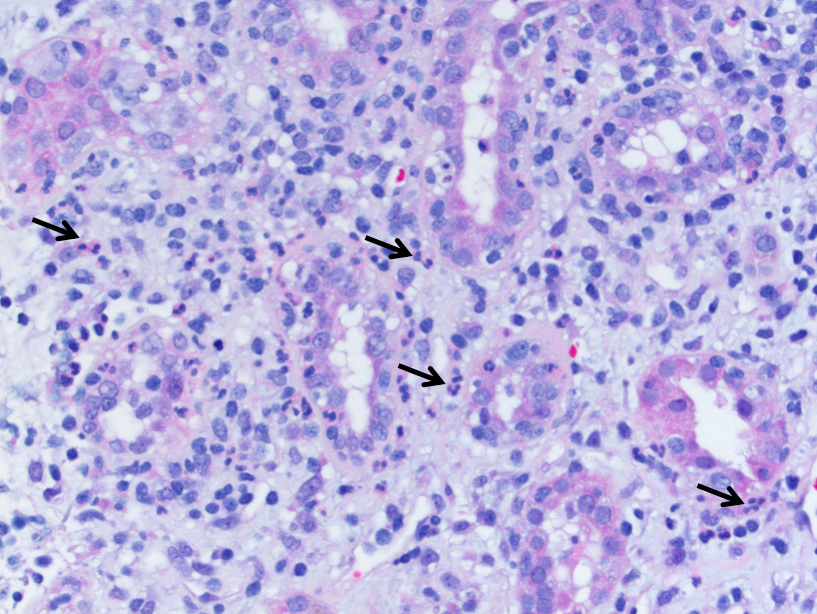
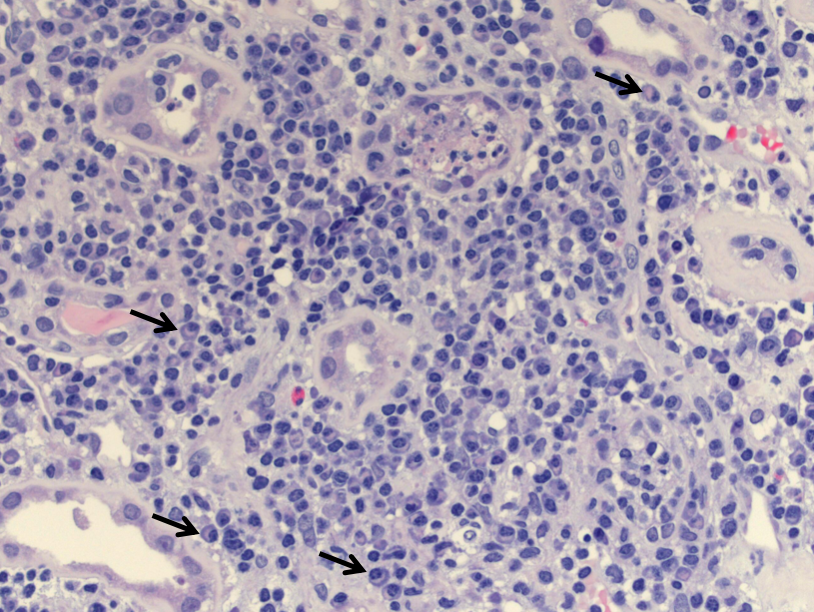
Viral infections of the kidney can also result in prominent interstitial inflammation, often in immunosuppressed individuals. HIV-associated nephropathy (HIVAN) is thought to be at least partly caused by direct infection of the kidney by the HIV virus. HIVAN affects all segments of the nephron and is characterized by collapsing glomerulopathy, microcystic tubular dilation, and tubulointerstitial nephritis. The interstitial nephritis that accompanies HIVAN can be predominantly plasmacytic in nature, and is also thought to be driven by the HIV virus as it typically improves with commencement of HAART. The immunocompromised state of HIV patients puts them at increased risk for other infections that cause AIN including polyomavirus, adenovirus, CMV, and EBV. Other unusual immunologic syndromes that can cause AIN in HIV patients include immune restoration inflammatory syndrome (IRIS) and diffuse infiltrative lymphocytosis syndrome (DILS).
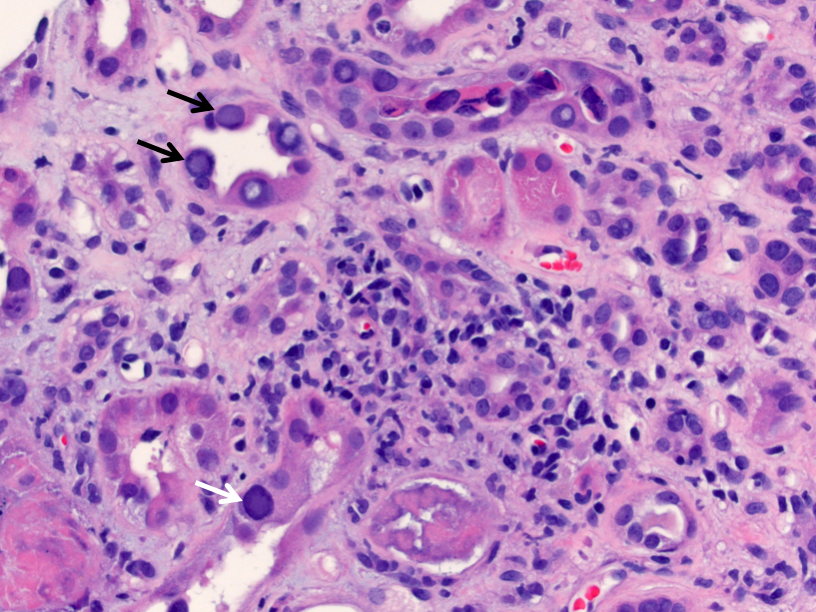
Polyomavirus infection can occur in immunosuppressed or immunocompromised individuals, with ~5% prevalence in kidney transplant patients. Polyomavirus nephropathy, usually caused by BK virus, is characterized by lymphoplasmacytic interstitial inflammation and tubulitis which preferentially involves the distal nephron segments. The tubular epithelial cells can show viral cytopathic effect with enlarged, hyperchromatic nuclei and “ground-glass” intranuclear inclusions. Immunohistochemistry for SV40 large T antigen confirms the diagnosis.
Autoimmune Disease or Immune Dysregulation
Autoimmune AIN is usually associated with systemic diseases such as systemic lupus erythematosus (SLE), sarcoidosis, Sjogren syndrome, tubulointerstitial nephritis with uveitis (TINU) syndrome, and IgG4-related disease. Approximately 50% of biopsies in SLE patients show lymphoplasmacytic tubulointerstitial nephritis, occasionally with lymphoid follicle formation. The extent of interstitial inflammation in SLE often correlates with the presence and extent of tubulointerstitial immune complex deposition, detectable by immunofluorescence microscopy.
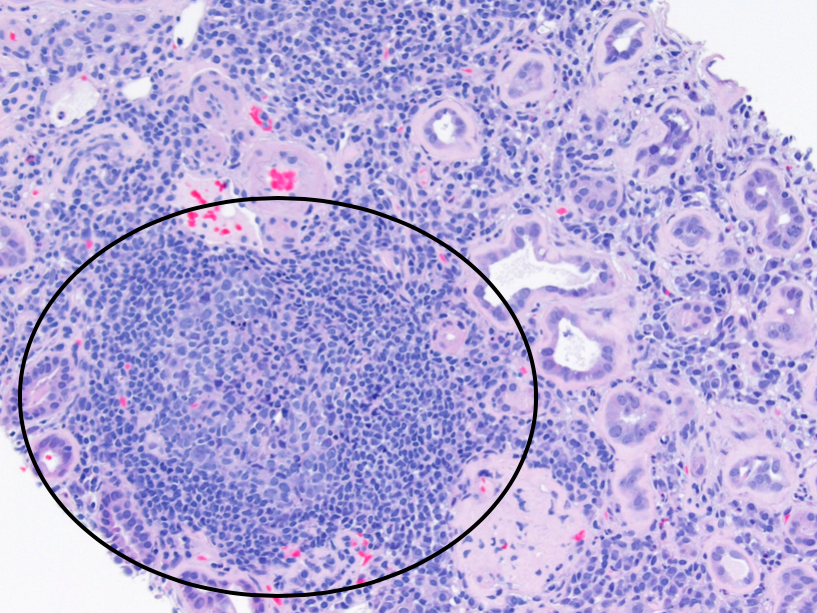
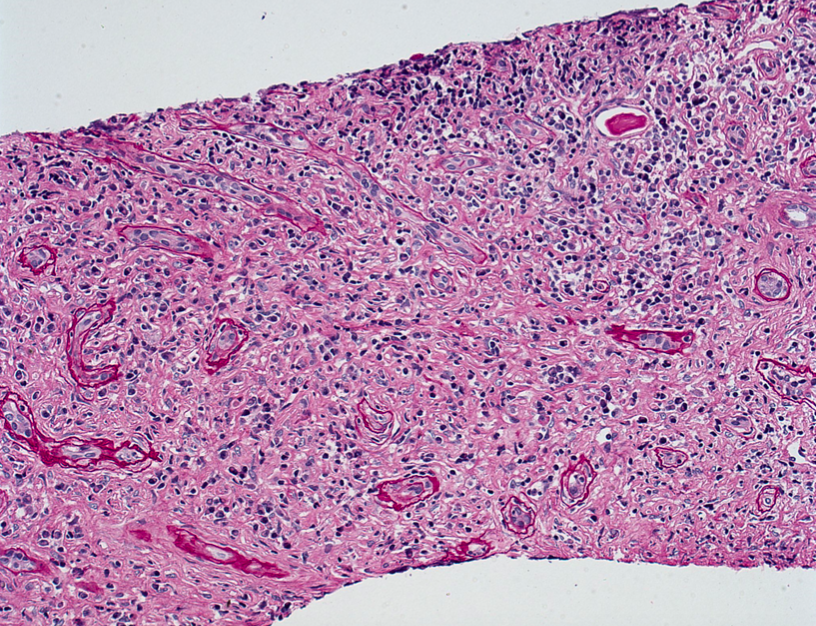
Lymphoplasmacytic tubulointerstitial nephritis is also characteristic of Sjogren syndrome. Sarcoidosis may show AIN with nonnecrotizing granulomatous inflammation. TINU is a pediatric disease characterized by AIN with a favorable course and uveitis with a chronic relapsing course. TINU can show a mixed interstitial infiltrate that closely mimics drug-induced AIN, and can also be granulomatous. IgG4-related disease is a systemic fibroinflammatory disease that can affect nearly any organ. IgG4-related interstitial nephritis is characterized by a plasma cell-rich interstitial infiltrate with increased IgG4+ plasma cells demonstrated by immunohistochemical staining, and granular tubular basement membrane immune complex deposits by immunofluorescence microscopy. As the disease progresses, there is characteristic expansile interstitial fibrosis in the renal cortex with a storiform pattern.
Kammi J. Henriksen, MD
Associate Professor of Pathology, University of Chicago