The kidney is all about balance; specifically the balance of fluid and electrolytes. This is in the face of sometimes an incredible onslaught of perturbations in homeostasis. To accomplish this impressive feat, the kidney is equipped with a complicated network of glomeruli, differentiated tubule segments, and intertwined capillaries. In order to achieve balance, the human kidney has developed two important feedback mechanisms to help autoregulate and maintain the integrity of this system. These two mechanisms, in which the macula densa and arterioles are key players, are called tubuloglomerular feedback (TGF) and the myogenic response (Figure 1). These feedback systems work together in the way that output controls the input in a loop. It can be difficult to parse these mechanisms of homeostasis, but understanding these processes will aid in the design of experiments and interpretation of results when looking into renal physiology and pathological mechanisms in disease states. TGF classically is understood to protect balance in the kidney by responding to increased sodium chloride (NaCl) delivery at the macula densa, where the glomerular capillary hydrostatic pressure and single nephron glomerular filtration rate (GFR) are reduced by a preglomerular vasoconstrictive response (Welch and Wilcox, 1997). Therefore, TGF regulates sodium excretion through the modulation of GFR, affecting sodium delivery (Welch and Wilcox, 1997)). GFR is responsible for determining the amount of fluid flowing through the tubules and is affected by many different variables, including changes in oncotic pressure and capillary surface area (Romero and Carretero, 2019). The afferent arteriole delivers blood to the glomerulus, which is brought into contact with specialized cells in the distal tubule, known as the macula densa. These areas of contact facilitate the crosstalk involved in these feedback mechanisms.
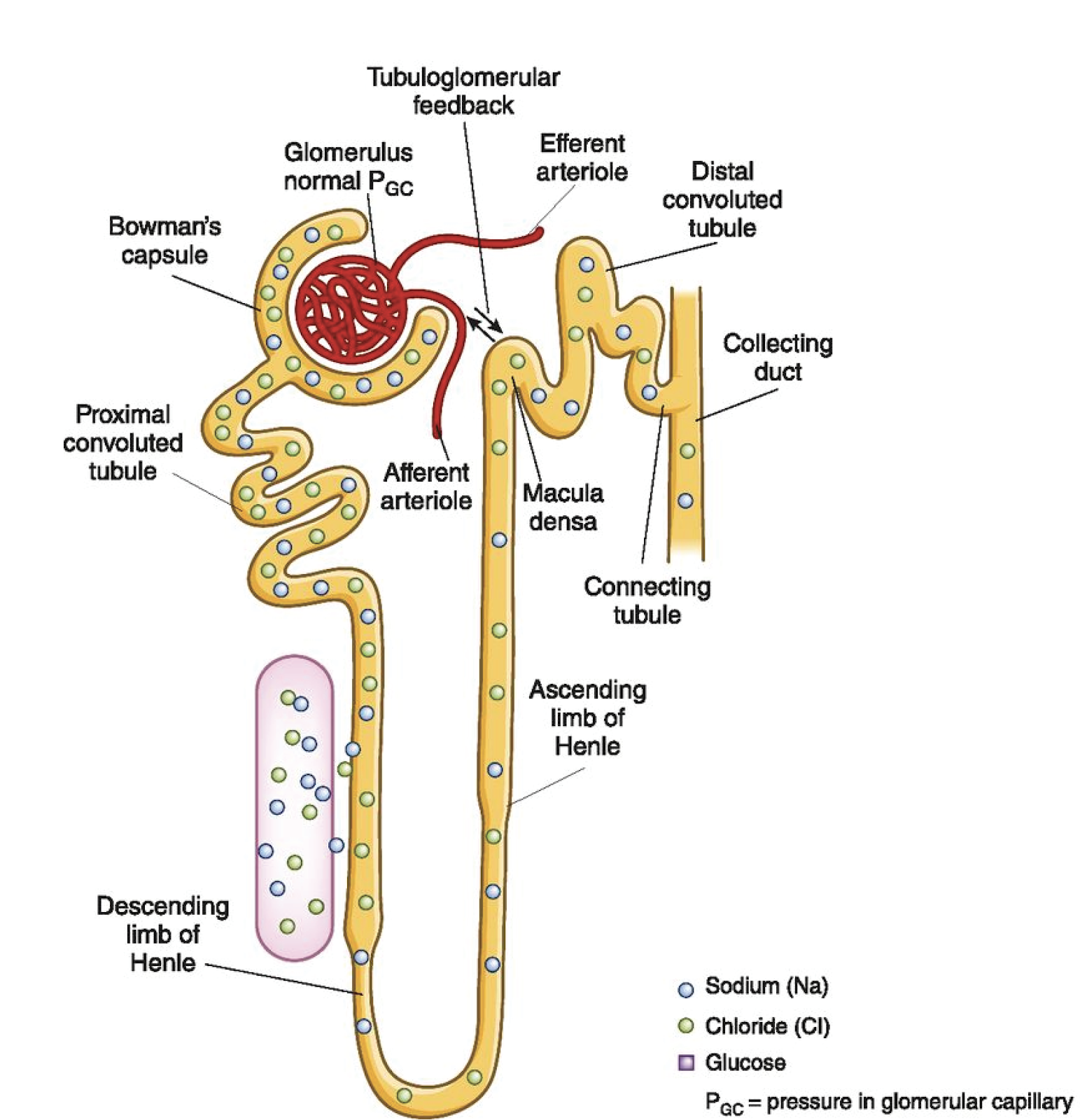
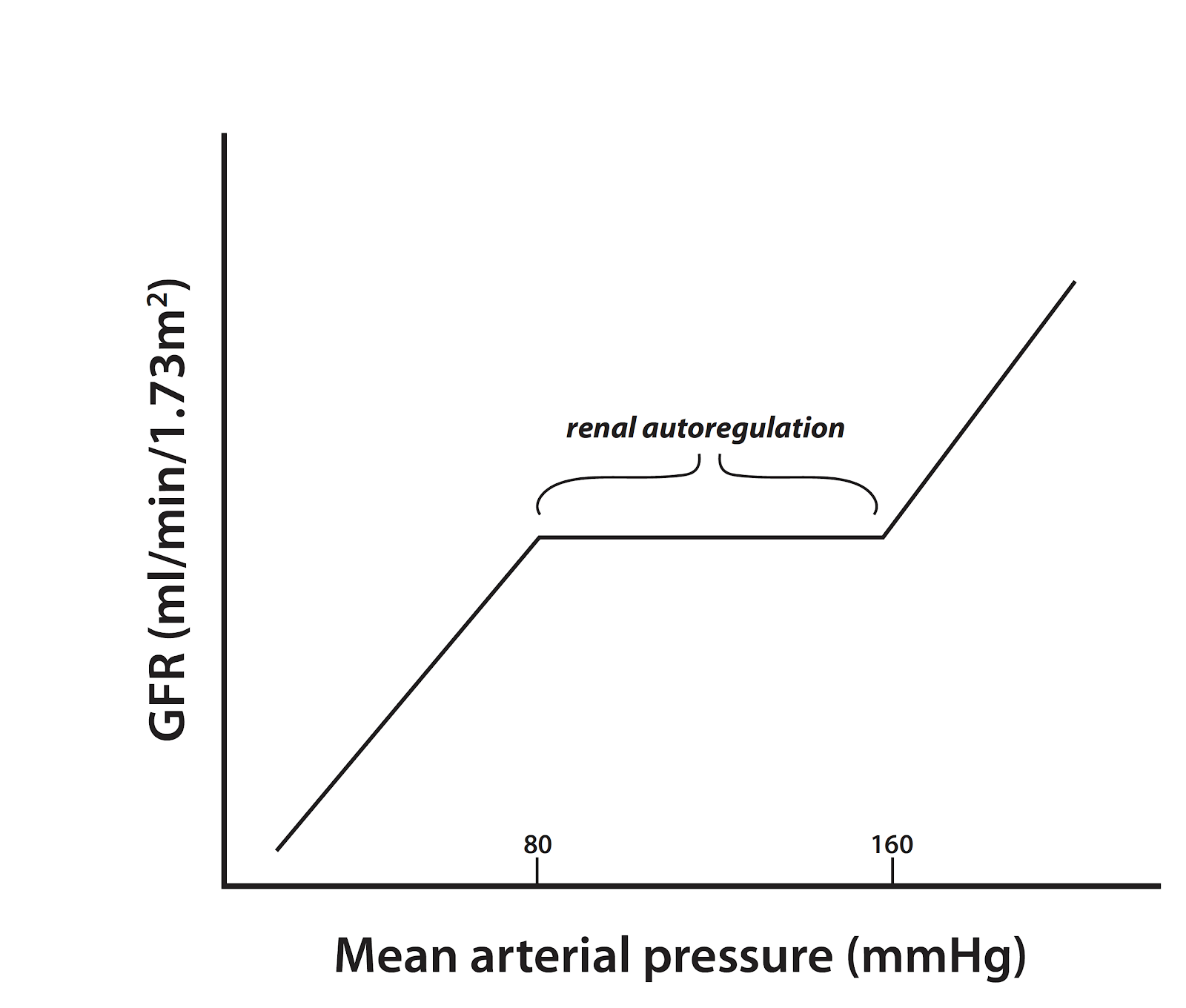
The kidneys employ these feedback mechanisms in order to “autoregulate”, which is the ability to maintain renal blood flow and GFR along a range of kidney perfusion pressures (Figure 2). Particularly in disease, blood flow and glomerular pressure can be disrupted, which has detrimental consequences on the function of the kidneys, potentially leading to long term, permanent damage of this specialized capillary network (Romero and Carretero, 2019). A recent paper from Zhang et al in JASN demonstrates in early diabetes that neuronal nitric oxide synthase (NOS1) may play a critical role in the TGF response, as well as control of GFR, sodium excretion, and blood pressure (BP) using macula densa-specific NOS1 knockout mice and isolated juxtaglomerular apparatus (Zhang et al, 2019). In the macula densa, NOS1 is the main nitric oxide (NO) synthase and negatively regulates TGF by blunting responsiveness via NO-mediated dilation of the afferent arteriole. The potential for nitric oxide (NO) in diabetic hypertension has been previously explored, but this group took the investigation a step further by suggesting a signaling relationship between the sodium-glucose cotransporter 1 (SGLT1) and NOS1 in hyperglycemia-induced glomerular hyperfiltration (Zhang et al, 2019). While this paper focused on the specific mechanism dependent on SGLT1, there is a discussion of the additive role of SGLT2 mediating sodium-glucose reabsorption to decrease NaCl delivery during hyperglycemia, and how both SGLT1 and SGLT2 contribute to glomerular hyperfiltration in diabetic patients.
Understanding the regulation of TGF through these pathways could provide great insight into the hyperfiltration associated with diabetes (Zhang et al, 2019). SGLT1 is found in the late segment (S3) of the proximal tubule, responsible for glucose resorption. In healthy states or normal glycemic conditions, there is little luminal glucose that reaches the macula densa. However, in hyperglycemia where the proximal tubule cannot completely reabsorb additional glucose, distal delivery is increased. To help clarify how the macula densa responds to increased glucose under these disease states, Zhang et al. showed that changes specifically in luminal D-glucose enhanced macula densa NO production. Use of a selective SGLT1 inhibitor, KGA-2727, on isolated JGA from mouse kidney reduced glucose-induced macula densa NO generation, demonstrating that increases of glucose at the MD inhibits TGF and stimulates NO generation via SGLT1. These findings further supports a relationship between SGLT1, NO, and changes to TGF (Figure 3).
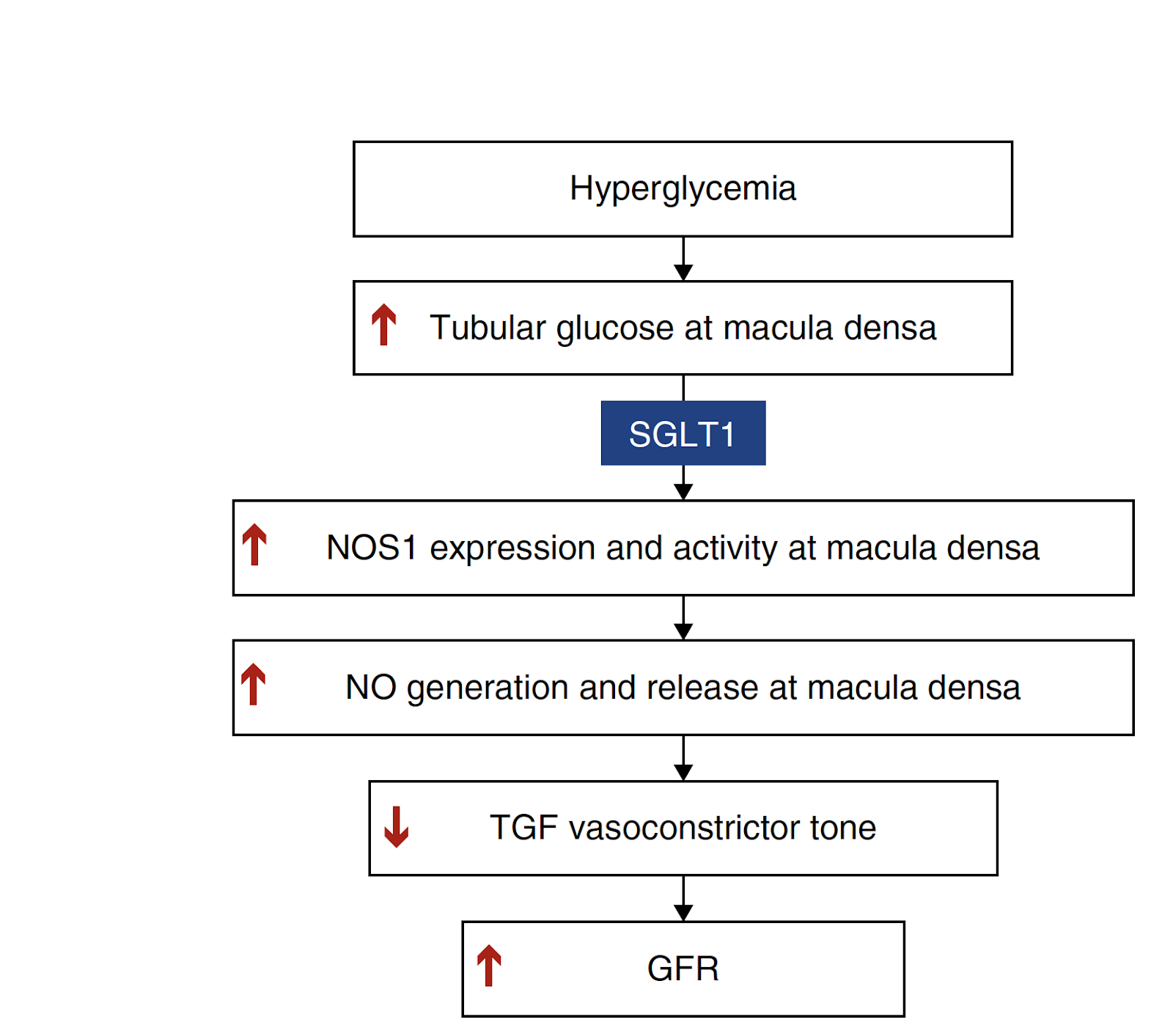
However, while intriguing, SGLT1-NOS1 is just part of the story, working additively with SGLT2-NaCl pathways to alter vasoconstrictor TGF tone and GFR in diabetic kidneys. Single-cell RNA-sequencing revealed that, in addition to SGLT1, GLUT1 is a robustly expressed glucose transporter in the macula densa. GLUT1 also plays a role in intracellular glucose efflux on the basolateral membrane of proximal tubule epithelial cells and has been implicated in stimulating NO production following dilation of afferent arterioles after increases in luminal glucose. The SGLT1-NOS1-TGF pathway defined here by Zhang et al has opened the door for further understanding of diabetic hyperfiltration. However, the dynamics of TGF regulation by the interplay of SGLT-NOS1, SGLT2-NaCl, and GLUT1, as well as the difficulties in targeting SGLT1 due to its expression in the gut, making it critical to define this very prominent and dire pathogenic mechanism (Figure 3).
Eryn E. Dixon
PhD Student
University of Maryland
Interesting fact: The first SGLT1/2 inhibitors (flozins) were isolated from the bark of apple trees

