Hyperoxaluria can cause a spectrum of disease ranging from urinary stone disease (USD), chronic kidney disease (CKD), end stage kidney disease (ESKD) to systemic deposition of oxalate leading to end organ damage. It can be broadly divided into primary disease, associated with increased production versus secondary or enteric hyperoxaluria due to increased absorption of oxalate from the gut.
The primary hyperoxalurias are inherited in an autosomal recessive fashion and divided into three types based on the different responsible genes: type 1 (AGXT, alanine glyoxylate aminotransferase), type 2 (GRHPR, glyoxylate and hydroxypyruvate reductase) and type 3 (HOGA1, 4-hydroxy-2-oxoglutarate aldolase).

Primary hyperoxaluria type 1 is a rare inborn error of glyoxylate metabolism which can present with kidney injury and systemic oxalosis in childhood. It has a prevalence of 1-3 per million. It results from mutation of the AGXT gene which codes for alanine glyoxylate aminotransferase (AGT), which normally converts glyoxylate into glycine. The video above illustrates the mechanism of oxalate generation in the absence of this enzyme. In the absence of AGT, glyoxylate is converted to oxalate.

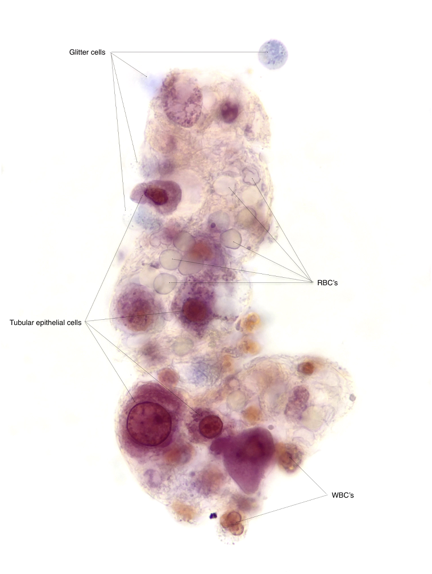
As oxalate is excreted primarily by the kidneys, these patients may go on to develop oxalate nephropathy, nephrocalcinosis, nephrolithiasis leading to CKD and ESKD.

(Image adapted)
With worsening kidney function due to oxalate nephropathy, systemic oxalosis occurs in the setting of increasing plasma levels of oxalate which the kidney is unable to excrete. At the level of 30 μ/L plasma supersaturation can occur leading to tissue deposition.

Case: Patient is a 43-year-old man with a history of nephrolithiasis since childhood, who developed ESKD at age 43 and started hemodialysis (HD). He underwent genetic testing at the time of pre-transplant evaluation and was found to have two pathogenic variants of AGXT gene confirming a diagnosis of primary hyperoxaluria (PH) type 1.
There are currently no approved drugs for the treatment of PH. Existing treatment options includes maintaining fluid intake and use of citrate supplementation to correct hypocitraturia to decrease oxalate supersaturation in urine. Pyridoxine, a cofactor for the defective AGT enzyme, can reduce the endogenous oxalate production in about one-third of patients with PH type 1. Restricting intake of vitamin C and oxalate containing foods forms the other arm of therapy. In patients with PH type 1 and ESKD, simultaneous liver-kidney transplantation or two- timed liver kidney transplantation, is recommended as HD is not sufficient to decrease the oxalate burden.
Lumasiran is an RNAi therapeutic that is currently in a Phase 3 (ILLUMINATE) trial that silences the gene that encodes for glycolate oxidase and prevents the generation of glyoxylate, the substrate for oxalate production. Thus, it decreases oxalate generation and hyperoxaluria. It has shown promising results and may become a viable therapeutic option, preventing the need for organ transplantation and complications of systemic oxalosis.
With no approved drugs at this time, simultaneous liver-kidney transplantation was recommended for our patient, as it is the standard of care for patients presenting with ESKD by preventing recurrent oxalate nephropathy in the allograft kidney by providing the missing enzyme. His pre-operative level of oxalate in plasma was 104 μ/L (normal is <5). It is important to decrease rebound hyperoxaluria that occurs in the peri-operative period (due to increased total body oxalate from systemic deposition) to prevent recurrent oxalate nephropathy – especially in the setting of delayed or slow graft function.

We used a combination of Continuous Renal Replacement Therapy (CRRT) and HD in the immediate post-transplant period with close monitoring of plasma oxalate levels to maintain low oxalate levels. HD was completely discontinued on post-operative day 15 as the patient demonstrated excellent allograft function and oxalate levels remained stable at around 10 μ/L.
Patient continues to do well and has a creatinine of 0.9 mg/dL, 90 days after combined liver-kidney transplant.
Post by: Sam Krishnamoorthy, MD
Nephrology Fellow, Washington University at St. Louis