Welcome to the 14th case of the Skeleton Key Group, a team of 40 – odd nephrology fellows who work together to build a monthly education package for the Renal Fellow Network. The cases are actual cases (without patient identifying information) that intrigued the treating fellow.
Hello nephro nerds! This time we bring you a baby case (diaper alert!) of hyponatremia and hyperkalemia! Let’s see if it intrigues you as much. Join us as we take you through the amazing world of Pediatric Nephrology
Written by: Caoimhe Costigan and S. Sudha Mannemuddhu
Visual Abstract: Narjes Alamri
A. The Stem
A 20-day-old male breastfed infant presented to the emergency department with poor feeding. On presentation, the child’s mother reported that he had been unwell for three days with decreased interest in feeding, a few episodes of small amounts of vomiting, less wakefulness and a weak cry. There was no history of fever, or trauma.
Patient was born at term by spontaneous vaginal delivery with a birth weight of 3.4 kg (appropriate for gestational age at 39 weeks) following an uncomplicated pregnancy. He was the second child of non-consanguineous parents. Newborn metabolic screening (for inborn errors of metabolism, cystic fibrosis and hemoglobinopathies) and hearing assessment were unremarkable.
Growth:
The infant’s weight was 3.3 kg. Although it is normal to lose weight following delivery, a well infant is expected to return to birth weight or higher by approximately 14 days of age.
This infant was still 100 g below birth weight at 20 days.
Vital Signs (with Normal Values):
BP 82/50 mm Hg (60-90/45-65), HR 180 (110-180), RR 50/minute (30-50), SpO2 96% (95-100%), Temp: 37.4C (36.5-37.5)
Physical examination:
The infant was pale and mottled. Central capillary refill time was prolonged at 4 seconds, and peripheries were cool. Anterior fontanelle was sunken. Heart sounds were normal without murmur, and lung fields clear on auscultation. Femoral pulses were present.
The infant was drowsy and intermittantly moaning. Primitive reflexes were intact and no focal neurologic deficits were detected.
Abdominal examination was unremarkable. Normal male genitalia with bilateral descended testes was noted. No rashes or abnormal pigmentation was noted.
Current Medications: None
B. The Labs
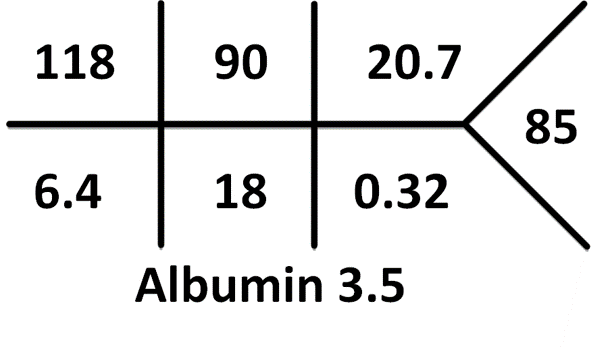
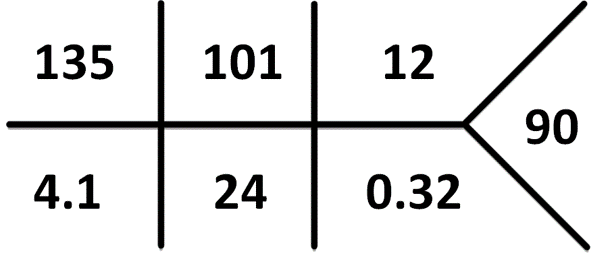
Normal lab values for age can be found here. Normal Creatinine in this age group is 0.2-0.4 mg/dL. Remember: Pseudohyperkalemia is a common cause of hyperkalemia in infants and children due to the nature of drawing blood from a heel stick or a small bore intravenous line leading to hemolysis – always consider repeating the sample if suspicious.
[UK values: pCO2 5.4 kPa]
What is the acid-base disorder?
Always remember to calculate the anion gap
Na+ – (Cl– + HCO3–) = 118 – (90 + 18) = 10 This is a normal anion gap metabolic acidosis.
Serum Osmolality was 263 mOsm/Kg (Normal 275-290 mOsm/Kg)
C. More Data
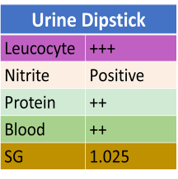
Microscopy shows >1000 WBC/hpf, >100 RBC/hpf and subsequently a pure growth of a pan sensitive E. Coli grows (>100,000 CFU/ml).
Urine electrolytes were unfortunately not obtained prior to fluid resuscitation.
The infant went on to have a renal ultrasound which demonstrated bilateral hydroureteronephrosis. Subsequent voiding cystourethrogram confirmed bilateral Vesico-Ureteral Reflux.
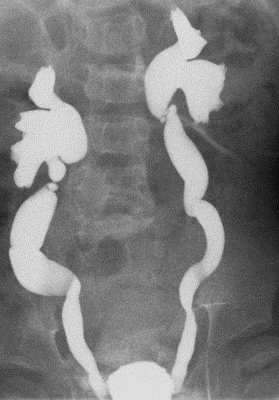
Source: Radiopedia
D. Differential Diagnosis
The biochemical constellation of hyponatremia, hyperkalemia and NAGMA suggests hypoaldosteronism. Disruption of the action of aldosterone could occur anywhere from production in the adrenal gland to its action at the collecting duct.
Let’s review the physiological mechanism that could lead to this biochemical picture.
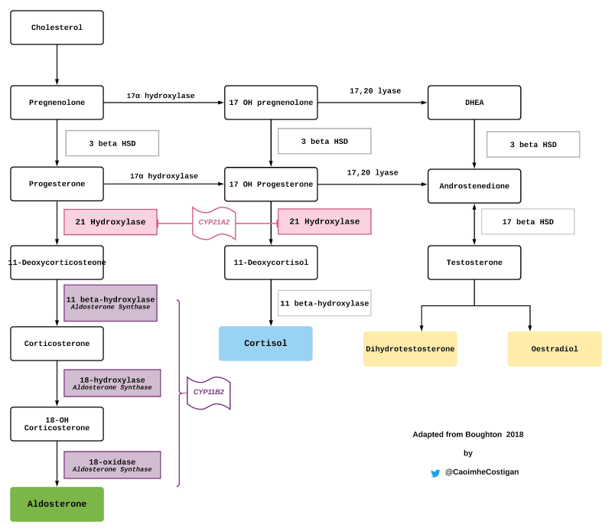
Aldosterone is a mineralocorticoid hormone produced in the adrenal cortex. Aldosterone passively crosses the epithelial membrane and interacts with the intracytoplasmic mineralocorticoid receptor in the principal cell at the cortical collecting duct (Fig 2)
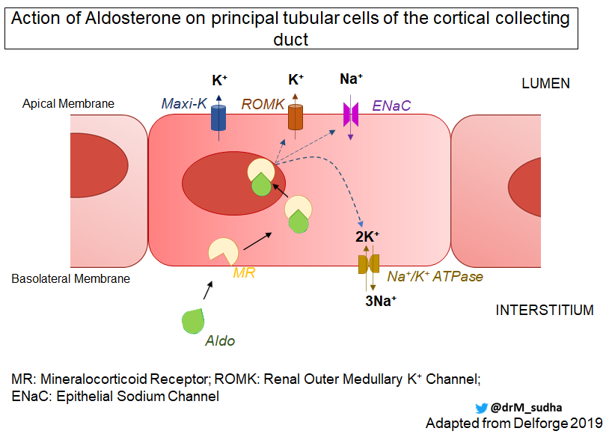
The aldosterone/MR complex translocates to the cell nucleus where it induces/represses gene transcription and via a complex series of interactions has the following effects:
- Enhances sodium transport by increasing the open probability of Epithelial sodium channels (ENaC) on the apical membrane and increasing the number of active channels, as well as indirectly upregulating Na+/K+ ATPase.
- This upregulation of Na transport provides a favourable electrochemical gradient for K+ secretion (via ROMK and Maxi K). Aldosterone upregulates ROMK transcription to maintain this electrochemical gradient.
- H+ concentration decreases in serum as distal tubular H+ secretion increases by:
- increasing the activity of the H+ /K+ -ATPase luminal pump on intercalated cells
- generating a lumen-negative potential by ENaC, favoring H+ secretion
What could cause this low aldosterone state in an infant?
It is helpful to consider two broad categories that could explain this presentation.
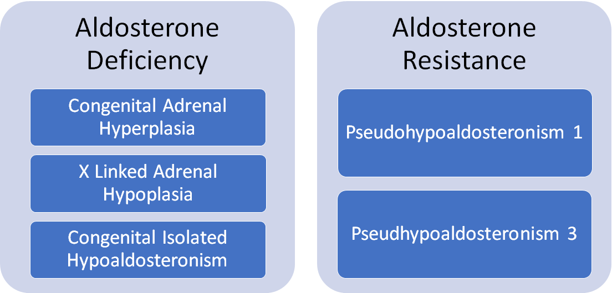
- Aldosterone Deficiency

- CAH is a group of inherited disorders of adrenal steroidogenesis
Epidemiology
- >95% of cases of CAH are due to deficiency of the enzyme 21-hydroxylase (21-OH)
- ~1 in 15,000 live births (variation in different ethnic groups)
Pathophysiology
- 21-OH converts 17-hydroxyprogesterone (17-OHP) to 11-deoxycortisol a key step in cortisol synthesis (fig 2)
- ↓ cortisol production → increased CRH/ACTH secretion (loss of negative feedback) → adrenal stimulation (hypertrophy)→ increased androgen production → virilisation
- Melanocyte Stimulating Hormone (MSH) is co-secreted with CRH which explains the pigmentation sometimes seen in this condition
- If enzyme activity is sufficiently impaired aldosterone synthesis is also disrupted
Genetics
- 21OH is encoded by the gene CYP21A2 – located on chromosome 6p21.3
- The pattern of inheritance is Autosomal Recessive
- Other less common genetic mutation which can cause CAH include:
o CYP11B1→ 11 𝛽 Hydroxylase Deficiency (1:100,000)
o CYP17→ 17 α Hydroxylase Deficiency (Rare)
o HSD3B2 → 3 beta Hydroxysteroid Dehydrogenase Deficiency (Rare)
o StAR → cholesterol transport defect
Clinical Presentation
- 21OH deficiency (21-OHD) is defined as “Classical” (severe) and “Non-Classical” (mild) forms depending on the severity of the enzyme deficiency
- ~2/3 of children with Classical 21OHD CAH also have aldosterone deficiency – “Classical Salt-Losing” form, with 1/3 having a “Simple-Virilising form”
- Females with classical CAH are generally detected early with ambiguous genitalia
- Males with classical salt-losing CAH present within the first few weeks of life with hypovolaemia, hyponatremia and hyperkalemia, those with classical simple-virilising present later with early virilisation (tall stature, early pubic hair, body odour etc)
- Non Classical forms present later with precocious puberty, hirsutism, oligo or amenorrhoea
Diagnosis
- Elevated serum concentration of 17-OHP (the substrate of 21OH) confirms the diagnosis
- 21OHD CAH is part of the Newborn Screening Programme in many countries including most of the United States (measures 17-OHP)

Epidemiology
- This is a very rare condition (exact prevalence unknown)
Pathophysiology
- Aldosterone synthase is an mitochondrial cytochrome p450 enzyme found in the zona glomerulosa of the adrenal cortex
- Aldosterone synthase catalyzes the conversion of deoxycorticosterone to aldosterone which involves three steps; 11𝛽-hydroxylation, 18𝛽-hydroxylation & 18-oxidation
- Aldosterone synthase deficiency → hypoaldosteronism
- Differs from CAH as cortisol and sex hormone biosynthesis are unaffected
Genetics
- Aldosterone synthase is encoded by the gene CYP11B2 – located on chromosome 8q24.3
- Different mutations (frameshift to point mutations) lead to varying degrees of enzyme deficiency
- Autosomal dominant and recessive forms exist
Clinical Presentation
- Can vary as other adrenal steroids can have a mitigating mineralocorticoid effect
- Infants generally present with failure to thrive, volume depletion, hyponatremia and hyperkalemia
- The condition is better tolerated in adulthood when salt intake can be more readily regulated
Diagnosis
- Hyperreninaemic hypoaldosteronism with a salt losing clinical and biochemical picture and a normal cortisol response on ACTH stimulation test is suggestive
- Urinary Steroid Profile (USP) may be helpful
- Genetics are confirmatory

Epidemiology
- Adrenal hypoplasia congenita is a very rare condition (exact prevalence unknown)
Pathophysiology
- Mutation of NR0B1 gene a key regulator of development of steroidogenic tissue leads to abnormal adrenal development
Genetics
- NR0B1 gene (Nuclear Receptor Subfamily 0 Group B Member 1)
- X-linked pattern of inheritance
- Can present as part of a contiguous gene syndrome with glycerol kinase deficiency (GKD) and Duchenne’s Muscular Dystrophy
Clinical Presentation
- The age of onset of clinical manifestations and severity of this condition can vary; adrenal insufficiency presents in infancy in about 60% of cases
- Hypogonadotropic hypogonadism typically presents as delayed puberty, but can present as underdeveloped genitalia in infancy – this is variable in clinical presentation
Diagnosis
- Low plasma levels of all adrenal steroids (cortisol may be normal initially due to active production in the fetal cortex – but will decline)
- No response to ACTH stimulation testing
- Normal 17-OHP levels (different from CAH)
- Assessment of gonadotropins (LH & FSH) at the time of puberty (to detect hypogonadotropic hypogonadism
)
- Aldosterone Resistance

This is a rare group of inherited disorders characterized by kidney tubular unresponsiveness to aldosterone. PHA 1 can be sub-classified into two forms – systemic and kidney.
Epidemiology
- PHA 1 is rare
- Incidence is estimated at 1:47 000 live births (Systemic PHA 1:166,000 and Kidney PHA 1:66 000)
Pathophysiology
Systemic PHA 1
- A genetic mutation affecting the α/𝛽/γ subunits of ENaC leads to defective sodium transport
- The ENaC is expressed in multiple organs including the kidney, skin, colon, lungs, sweat and salivary glands – leading to a multi-organ or systemic defect in sodium transport
- This results in severe salt-wasting, volume depletion and persistent hyperreninaemic hyperaldosteronism
Kidney PHA1
- Sodium transport is affected via a disruption of aldosterone binding to the MR. Salt-wasting is generally less severe and limited to the kidney.
- Kidney PHA1 tends to improve with age – this may be related to increased MR expression with advancing age which may compensate
Genetics
Systemic PHA1:
- Loss of function mutation of the gene encoding the ENaC subunits – SCNN1A, SCNN1C and SCNN1G.
- The pattern of inheritance is autosomal recessive
Renal PHA1:
- Heterozygous mutation in the NR3C2 gene encoding the Mineralocorticoid Receptor (MR)
- >100 mutations have been identified with diverse clinical expression (nonsense & frameshift most common)
- The pattern of inheritance is autosomal dominant
Clinical Presentation
Systemic PHA:
- Infants present with severe salt-wasting, volume depletion with significant and often difficult to manage hyperkalemia, and metabolic acidosis
- Systemic features such as cholelithiasis, dermatitis, and respiratory symptoms can be part of the phenotype
Renal PHA1:
- Can also present as salt-wasting in infancy but tends to be less severe
Diagnosis
- Volume depletion,hyponatremia, hyperkalemia and metabolic acidosis with elevated renin and aldosterone
- Genetics to confirm the mutatio
n

This is a transient form of tubular aldosterone resistance (also known as PHA3)
Epidemiology
- Estimated 1 in 13,200
Pathophysiology
- Often presents in the context of urinary tract infection/pyelonephritis – particularly in infants with a structurally abnormal urinary system (such as vesicoureteric reflux or an obstructive uropathy)
- The exact mechanism of underlying aldosterone resistance in this context is not fully understood
- Tubular immaturity, inflammation and increased intrarenal pressure leading to parenchymal damage likely all play a role
Clinical Presentation
- Infants present with volume depletion,hyponatremia & hyperkalemia with urinary tract infection and/obstruction
- Can present acutely with signs of systemic illness/sepsis, or more insidiously with persistent failure to thrive
- Most infants are < 6 months although can be older, and occurs more frequently in boys than girls
Diagnosis
- Volume depletion,hyponatremia, hyperkalemia and metabolic acidosis with elevated renin and aldosterone
- Urine culture may demonstrate infection
- Ultrasound may demonstrate an abnormal urinary tract
A detailed review of PHA can be found here.
(Note: PHA2 is Gordon’s syndrome and not covered in this review)
A comprehensive review of “Salt-Wasting” in infancy can be found here and is summarized below:

How can you distinguish between different causes of “Salt-Losing” in infancy?
It can be difficult, particularly in an emergency situation. However it is important to distinguish between adrenal insufficiency and aldosterone resistance or isolated deficiency as they require different approaches.
Acute adrenal insufficiency is a medical emergency, and when suspected it is always appropriate to treat. Adequate fluid resuscitation and stress doses of hydrocortisone (100 mg/m2/day) should not be delayed while awaiting the outcome of definitive investigations. Obtaining a blood sample for analysis prior to initiation of glucocorticoid replacement, if possible, is very important in establishing a diagnosis.
Here’s the flowchart adapted by Manikam 2018 to aid in diagnosis:
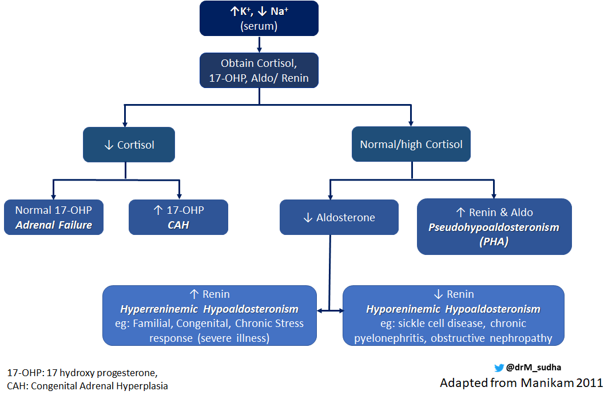
E. The Answer
Renin, aldosterone, cortisol, 17-hydroxyprogesterone (17-OHP) levels, and blood culture results returned on the patient confirming urosepsis with transient pseudohypoaldosteronism.
The diagnosis of urosepsis, with the biochemical picture of hyperaldosteronism and the ultrasound findings of bilateral hydroureteronephrosis lead us to this diagnosis.
A normal blood glucose level and normal blood pressure makes us less concerned about cortisol insufficiency as would be seen in CAH. The elevated plasma aldosterone level is consistent with pseudohypoaldosteronism. While we cannot rule out PHA 1 at this point, the findings of bilateral hydroureteronephrosis and urinary sepsis are particularly suspicious for transient PHA (PHA 3).
Transient PHA is usually related to infection in young infants with structural abnormalities of the urinary tract. Male infants are more often affected than females – perhaps relating to the higher incidence of obstructive uropathy in males. Bilateral malformations are at higher risk than unilateral, and VUR was the most common associated abnormality in one study. It is important to note that transient PHA can also occur in the absence of a structural abnormality – particularly in severe pyelonephritis. Transient PHA is often under recognised but may have a similar incidence to CAH.
The exact mechanism underlying transient PHA is not fully understood. Infants have very different kidney physiology to older children or adults; they have a lower GFR and their immature tubules have a reduced capacity for sodium and water reabsorption. Mineralocorticoid expression in the kidney is low at birth (despite high aldosterone levels) which may account for compromised sodium handling in this period. This baseline partial aldosterone resistance makes this age group particularly vulnerable to fluid and electrolyte abnormalities. In the context of infection or obstruction, inflammation and increased intrarenal pressure leading to parenchymal damage may exacerbate this immature tubular aldosterone resistance and account for the biochemical presentation seen acutely.
The electrolyte disturbance in transient PHA usually resolves with management of the underlying infection or alleviation of the obstruction.
Our patient received initial fluid resuscitation with normal saline bolus at 20 ml/kg bolus. This was followed by 0.9% saline and 5% dextrose infusion- rate titrated based on serial electrolyte measurement. Sodium was gradually corrected over 36 hours to prevent osmotic demyelination syndrome. For hyperkalemia, he received 10% calcium gluconate (0.1mmol/kg) and insulin-dextrose.
Empiric IV antibiotics were given. The infant clinically improved and his electrolytes normalized over the initial 36 hours of treatment. Normalisation of his electrolytes effectively rules out PHA 1, confirming the diagnosis of Transient PHA (PHA 3).
Following completion of his antibiotic course (14 days) he was discharged on Trimethoprim- Sulfamethoxazole as UTI prophylaxis given his diagnosis of VUR.
F. Take-Home Points
- When you see hyponatremia, hyperkalaemia, acidosis and salt-wasting – think hypoaldosteronism.
- Always remember – babies are not little adults! There are physiological differences – lower GFR, partial aldosterone resistance, and reduced tubular concentrating ability.
- The top differential for salt-wasting in infancy is usually CAH but it is not the only cause! Consider PHA 3 – transient tubular resistance to aldosterone seen in babies with urinary tract abnormalities (with or without infection) and in some cases of pyelonephritis with structurally normal urinary tracts.
- Management is to address the underlying trigger.



NICE ONE, THANK YOU