Oxalate is a terminal metabolite in humans. Glyoxylate becomes glycine and pyruvate via alanine-glyoxylate aminotransferase (AGT) action, and excess glyoxylate gets converted to oxalate by lactate dehydrogenase (LDH) and glycolate oxidase. Besides being derived endogenously through metabolism, oxalate can come exogenously from foods and beverages rich in oxalate such as spinach, beans, rhubarb and black tea. Exogenous ascorbic acid undergoes a degradation pathway with oxalate as a product.Oxalate in plasma is freely filtered by the glomerulus, then partially reabsorbed and secreted by tubular epithelium cells. Hyperoxaluria, defined as excessive urinary excretion of oxalate, is associated with kidney stone formation and even oxalate nephropathy. The deposition of calcium oxalate in the kidney can lead to progressive damage over time with the eventual development of chronic kidney disease (CKD). The putative mechanisms include tubular obstruction by crystals, interstitial inflammation, and tubular epithelial cell injury.
Clinical features of oxalate nephropathy
The primary hyperoxalurias (PHs) are genetic conditions that allow us to understand the role of oxalate in the development of kidney and systemic disease. PH are rare diseases due to mutation of genes encoding metabolic enzymes of glyoxylate metabolism, leading to overproduction of oxalate. PH commonly present in childhood, with disease spectrum extending from recurrent kidney stones, kidney oxalosis to end-stage kidney disease (ESKD). Systemic oxalosis usually appears after kidney damage, especially in the bones and blood vessel walls. Joints, retina, skin, bone marrow, heart and central nervous system can also get involved. In contrast, secondary hyperoxaluria (SH) is relatively more common with kidney dysfunction as a consequence. Secondary hyperoxaluria is usually caused by an increase in gut oxalate absorption, which is usually a consequence of increased dietary oxalate intake, intestinal surgeries (for example, Roux-en-Y gastric bypass), fat malabsorption (seen in conditions like pancreatic insufficiency and cystic fibrosis), or changes in the gut microbiome (for example, in the setting of oral antibiotic use). Other clinical variables associated with higher urinary oxalate excretion include age, higher BMI, and diabetes. Most patients present with acute kidney injury (AKI) or CKD. Laboratory investigations may show elevated 24-hour urinary oxalate excretion. Kidney biopsy is the definite diagnostic method and almost all patients had calcium oxalate deposition and tubulointerstitial injury.
Notably, in a retrospective cohort study of 615 biopsy specimens, the prevalence of oxalate deposition was 4.07% (25/615) and in 88% cases was a major finding. Oxalosis was anticipated in only 1 case prior to biopsy. The etiology was clarified retrospectively in 56% of cases with the rest remaining unknown. This finding suggests that kidney oxalate deposition is relatively common, but often unexpected on a kidney biopsy. Calcium oxalate may be the primary driver of kidney disease or superimposed on another process. However, patients with known and unknown etiologies showed no differences in clinical features, pathology or 3-month kidney survival rates. As prognosis data of secondary oxalate nephropathy from a meta analyses of case series and case reports, in 13 months mean duration follow-up, no patient experienced complete kidney recovery with 58% patients remaining dialysis-dependent. The overall mortality rate was 33%. Elevated creatinine at the time of biopsy, background chronicity change and higher calcium oxalate density are associated with ESKD.
Pathogenesis of oxalate nephropathy
Oxalate nephropathy could be in the form of AKI or CKD. The underlying mechanism of oxalate-induced kidney injury has been examined using in vivo and in vitro experiments. Mulay et al. described calcium oxalate crystals trigger kidney inflammation by nucleotide-binding oligomerization domain (NOD)-like receptor protein 3 (NLRP3)-mediated IL-1β secretion contributing to AKI. Their further work revealed the crystals also exert a direct cytotoxicity by inducing a receptor-interacting serine-threonine kinase-3 (RIPK3)-dependent and mixed lineage kinase domain-like (MLKL)-mediated necroptosis, which is regulated tubular cell necrosis. Their recent findings indicated that peptidylprolyl isomerase F (PPIF)-dependent mitochondrial permeability transition related necrosis and necroptosis contribute to the pathogenesis of oxalate-induced AKI. In contrast to oxalate-induced AKI, many recent studies have emphasized the inflammasome-independent functions of NLRP3 in the development and progression of CKD, for example, NLRP3-caspase-8 mediated epithelial cell apoptosis and TGF-β induced renal fibrosis.
Urine parameters in Diabetes Mellitus
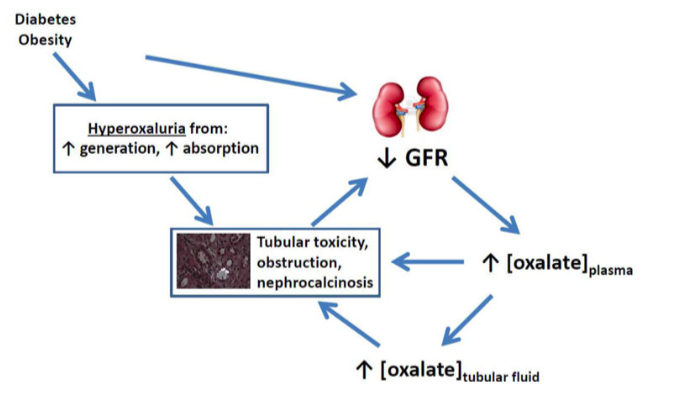
In a cross-sectional study of 3 large cohorts of Nurses’ Health Study (NHS) and Health Professionals Follow-up Study (HPFS), DM was found to be associated with increased risk of kidney stone formation. Prospective data of this study also demonstrated a history of kidney stones increases the likelihood of a subsequent diagnosis of T2DM over combined 44 years follow-up.Calcium oxalate is the most common composition of kidney stones. Large epidemiologic studies have shown the associations of urinary oxalate with diabetes. In a retrospective study of 1117 patients with nephrolithiasis, DM was associated with higher urine uric acid and oxalate, and lower pH.
Does urinary oxalate mediate kidney disease in Diabetes Mellitus
Plasma levels of glyoxylate and glyoxal, which are precursors of oxalate, were found to be higher in those with diabetes, even predating the diagnosis of DM by up to 3 years. Furthermore, studies using the Chronic Renal Insufficiency Cohort have found that higher 24-hour urinary oxalate excretion may be a risk factor in CKD progression and that those with diabetes had 11% higher urinary oxalate even after adjusting for relevant covariates. The metabolic features of oxalate in DM patients indicated a possible link between urinary oxalate and DM in renal impairment.
From a mechanistic standpoint, consistent evidence suggests that NLRP3 inflammasome in resident renal cells plays an important role in the progression of diabetic nephropathy. Furthermore, ROS induced NLRP3 inflammasome activation in podocytes results in podocyte injury and initiation of albuminuria, triggering diabetic nephropathy. Thus, researchers proposed that urinary oxalate may be a mediator of kidney disease in DM, as depicted in figure 1. Nevertheless, although there are links in metabolism and shared mechanisms, the interaction between oxalate and DM in the progression of CKD is overall understudied and warrants further investigation.
Summary
Emerging evidence indicates that oxalate precursors and urinary excretion are increased in DM patients, which may be associated with CKD progression. The NLRP3 inflammasome may play an important role both in DM and oxalate-related kidney injury. These pieces of evidence altogether imply urinary oxalate could mediate kidney disease in DM. However, in this largely unknown field, many puzzles need to be solved. What is the metabolic profile of oxalate in DM? By what mechanisms oxalate causes CKD in DM? Will oxalate lowering management be protective in diabetes and CKD?
Yani Zhang, MD
China Medical University
@YaniZhang7

