Washington University School of Medicine in St. Louis
Kidney cell type diversity, complexity, and interaction mechanisms are a source of intrigue and frustration for many. With more than 20 different, specialized cell types (Quaggin, 2019), elucidating the kidney’s cell specific physiological and pathophysiological mechanisms is a top priority shared by the field. Therefore, it is no surprise that a considerable portion of research efforts have been dedicated to bringing single cell sequencing technologies front and center as a tool for isolating cell types to uncover their individual functions. Let’s take a look at how this tool is specifically being used in research to explore sex differences in kidney disease. Epidemiological data has shown that different pathologies, such as polycystic kidney disease, IgA nephropathy, and of course, chronic kidney disease (CKD) exhibit sexual dimorphism, in other words different characteristics between males and females (Neugarten at al, 2000). Now, the question is how should we investigate the drivers of sex specific kidney physiology; the answer may lie in applications for RNA-sequencing.
The goal of RNA-sequencing is to develop an understanding of each cell type by profiling their identity using transcriptomics, followed by integration with other genomic, epigenomic, and proteomic methodologies (Stuart and Satija, 2019). Maybe the most discussed and praised of these technologies, single cell RNA sequencing (scRNA-seq) (Figure 1) leads the charge in illuminating transcriptional changes in disease, sex, and development. But, how one approaches RNA-sequencing can determine what experimental questions can be answered. For example, bulk RNA-sequencing, where each sequencing library represents a population of cells, evaluates average gene expression changes, while scRNA-sequencing reveals a gene’s expression across a single population of cells as each sequencing library is a single cell. Both can serve valuable purposes, but one approach may be more appropriate for the scientific question(s) posed. If you are interested in learning more about sequencing analyses, click the link for a great review by Stuart and Satija (2019).
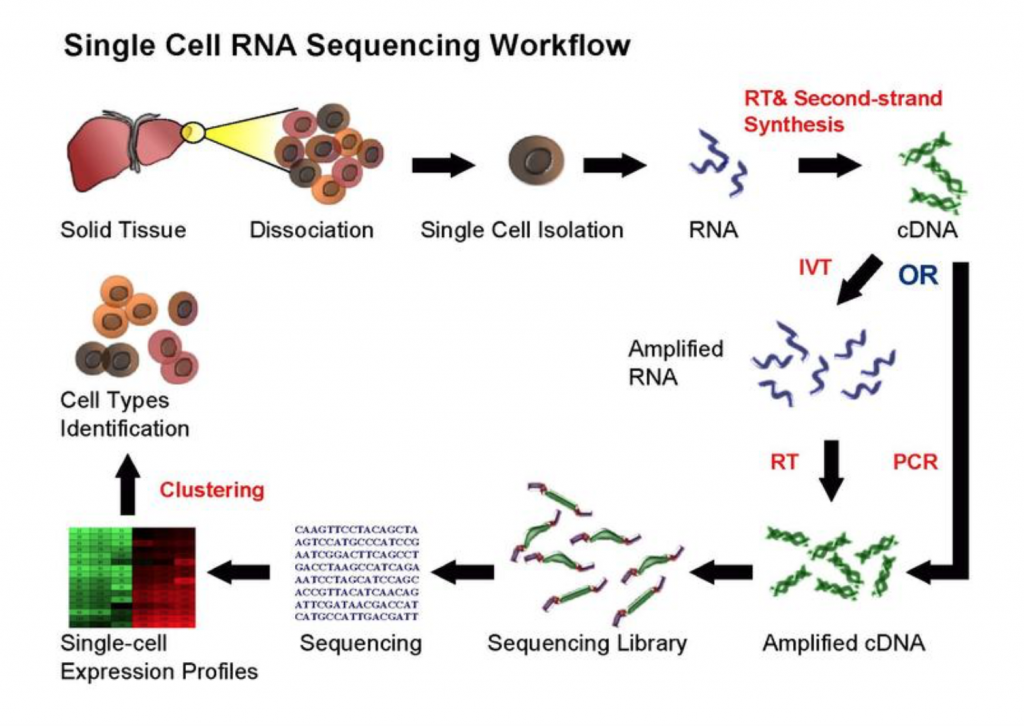
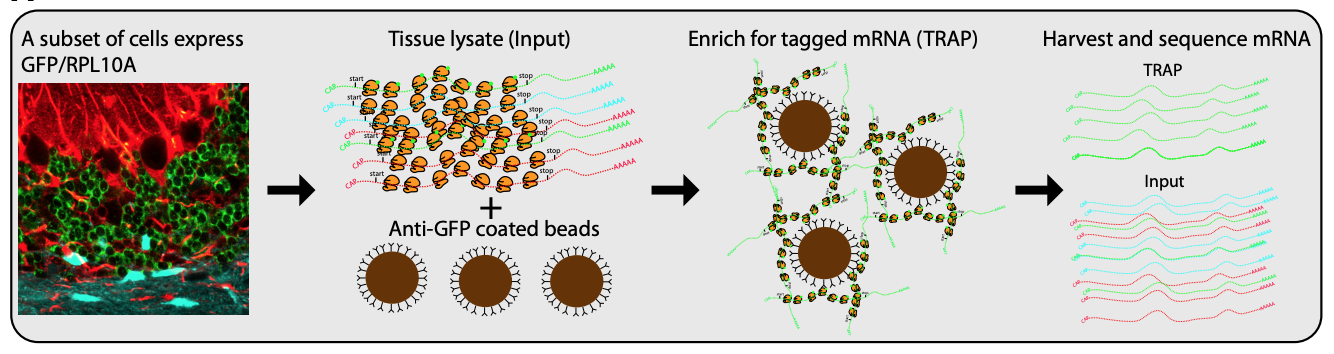
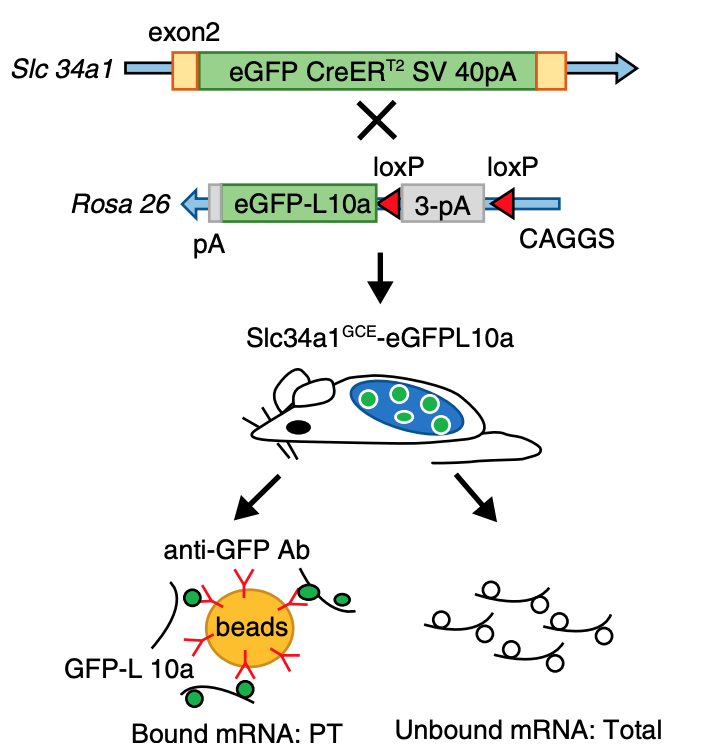
While single cell sequencing applications are taking the field by storm, there are drawbacks to solely using this approach to create transcriptional maps for kidney injury and disease. For example, these sequencing applications are very powerful in the identification of highly expressed genes, but can potentially miss lower/rarely expressed genes, which could turn out to be critical targets in physiological and disease mechanisms. Wu and Lai et al developed an approach to attempt to overcome this problem using a technique called translating ribosome affinity purification (TRAP) (Figure 2). TRAP is a tool that illuminates cell-specific transcriptional profiles, by sequencing mRNA that is bound by the ribosome while being translated. When compared to other sequencing methods, TRAP identifies more differentially expressed genes and can define regulatory genes missed by scRNA-seq due to its increased sensitivity for lower expressing genes (Wu and Lai et al, 2020). This technology works via inducing a particular cell type to make a L10a ribosomal subunit in which an eGFP-tag is placed. This L10a ribosomal subunit is part of the 60S subunit that catalyzes protein synthesis. The ribosomal subunit is then activated in a cell type of interest by using a promoter of interest.Affinity purification is applied to tissue lysate with eGFP antibodies to pull off mRNAs to be sent for sequencing (Liu et al, 2014). Wu and Lai et al applied this TRAP technology in male and female SLC34a1-eGFPL10a mice that have inducible expression of eGFP-L10a in the proximal tubule, driven by the SLC34a1 promoter (this is specific for differentiated epithelial stem cells in the proximal tubule)(Figure 3). The proximal tubule was the focus of these studies as this nephron segment is vulnerable to genetic, ischemic, and toxic insults attributed to its high metabolic activity. Furthermore, the proximal tubule has been previously shown to be enriched with sexually dimorphic genes (Ransick et al, 2019), making the proximal tubule an exciting and meaningful target for TRAP applications to study kidney function.
It has been suggested for decades, using in vivo model systems, that there may be protection elicited by female sex from progression to chronic kidney disease following injury (Neugarten and Golestaneh, 2019). However, the mechanisms for this are unknown. CKD is functionally defined by a reduction in functional kidney mass, and often features tubulointerstitial fibrosis, extracellular matrix deposition, expansion of myofibroblasts, tubular atrophy, and peritubular capillary rarefaction (Wu and Lai et al, 2020). The progression of kidney injury to CKD is hypothesized to be driven by the upregulation of proinflammatory and profibrotic pathways (Kirita et al, 2020). Currently, there is no way to reverse kidney disease, but there is an immediate call for better therapies as the burden of kidney disease continues to rise across the world (Pavkov et al, 2018). Understanding these disease mechanisms in men and women will lead to identification of new targets for treatment and management of CKD.
Wu and Lai et al decided to employ proximal tubule specific profiling with TRAP and cortex bulk RNA-sequencing to investigate sex dependent transcriptional signatures in healthy proximal tubule, as well as in inflammation and fibrosis following unilateral ureteral obstruction (UUO) surgery in male and female Slc34a1-eGFPL10a mice. Interestingly, a majority of the differentially expressed genes found between male and female mice in healthy proximal tubules were located in the autosomes, not the sex chromosomes. Further, Wu and Lai et al used RNA in situ hybridization (ISH) to visualize and confirm sex dependent expression of genes such as Hao2 (upregulated in females) and Cndp2 (upregulated in males). A large percentage (64.8%) of the sexually dimorphic genes were differentially expressed in males and females following UUO. Thus, proximal tubules of males and females have different translatomes under both healthy and injured states.
However, an unexpected and noteworthy finding of this study was the ability of TRAP to identify the differential expression of many non-protein coding transcribed RNAs, called long non-coding RNAs (lncRNAs). Not much is currently known about the function of lncRNAs in general, but the identification of differential lncRNA expression in UUO supports future investigation into their contribution to transcription and translation in disease progression. With sex specific mechanisms potentially involving numerous pathways in multiple cell types from the epithelia to the stroma to immune cells, it is becoming increasingly apparent that combinations and novel applications of sequencing approaches could present a golden opportunity to identify new and effective therapeutic targets.
Reviewed by: Elinor Mannon, Kelly Hyndman, PhD, Matthew Sparks, MD, FASN, FAHA