Welcome to the 31st case of the Skeleton Key Group, a team of nephrology fellows who work together to build a monthly education package for Renal Fellow Network.
Authors: Russell Leong, Trey Richardson
A. The Stem
A 41-year-old man with alcohol use disorder and chronic hypokalemia of unclear etiology on potassium supplementation presents to the emergency department with bilateral lower extremity weakness. Two weeks ago, he presented with lower back pain and was treated with a course of steroids. Labs at that time were unremarkable, including a normal potassium of 4.8 mEq/L. He was instructed to discontinue his potassium supplements. In the following days, he developed random episodic sudden onset lower and upper extremity weakness that progressed to lower extremity paralysis on the day of presentation.
During triage, he developed sustained supraventricular tachycardia (SVT) that progressed into pulseless polymorphic ventricular tachycardia (VT). Return of spontaneous circulation was achieved after 6 minutes of ACLS.
Post-Arrest Examination
Vitals: Temp 97.6°F, BP 93/54 mm Hg, HR 88 bpm, intubated and sedated.
General appearance: intubated, sedated adult male.
CV: normal rate and regular rhythm. 3/6 systolic ejection murmur.
Lungs: mechanical breath sounds bilaterally.
Neurologic: GCS 3, sedated on propofol. PERRL.
Remainder of the exam was within normal limits.
B. The Labs
Initial Labs (Pre-Arrest):
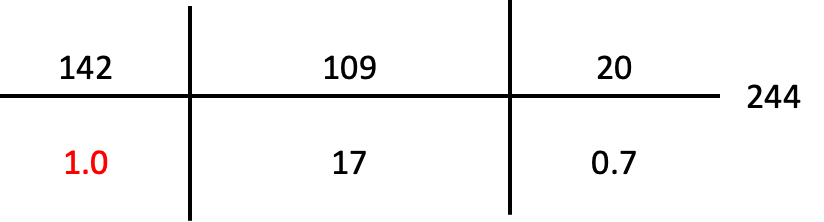
Calcium 10.2 mg/dL
Magnesium 1.9 mEq/L
Total bilirubin 0.8 mg/dL
Hemoglobin 15 g/dL
Ethanol negative.
TFTs: TSH undetectable, FT4 1.77 ng/dL
Post-Arrest Labs:
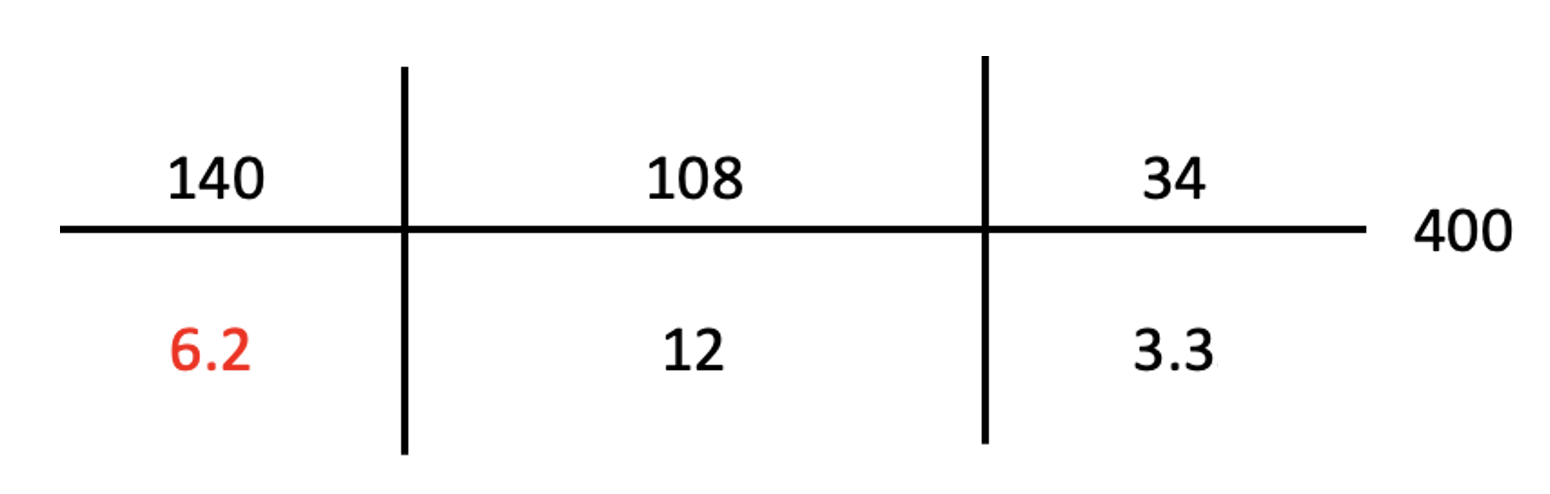
Calcium 7.9 mg/dL
Magnesium 2.7 mEq/L
Hemoglobin 11.9 g/dL
Anion gap 20
ABG: pH 7.15/pCO2 38/pO2 198/ Bicarbonate 12 consistent with a non-anion gap, anion gap metabolic acidosis, and respiratory acidosis.
Lactic acid 4.3 mEq/L
CXR showed endotracheal tube in the appropriate position without evidence of pleural effusions, infiltrates, or pulmonary edema
C. Differential Diagnosis for Hypokalemia
Our patient presented with severe manifestations of hypokalemia including skeletal muscle paralysis, glucose intolerance, and cardiac arrhythmias in the form of SVT and VT.
It is helpful to divide the causes of hypokalemia into three main categories: redistribution, non-kidney losses, and kidney losses as seen in the table below.
| Redistribution | Non-renal losses | Renal losses |
| Beta agonist administration | GI losses* | Diuretic use |
| Insulin administration | Hypomagnesemia | |
| Alkalemia (e.g., bicarbonate administration) | Hyperaldosteronism | |
| Thyrotoxic and hypokalemic Periodic paralysis | Proximal and distal RTA | |
| Barium toxicity | Metabolic alkalosis bicarbonaturia | |
| Treatment of B12 deficiency (rapid cell synthesis) | Genetic syndromes: Bartter syndrome, Gitelman syndrome, Liddle syndrome | |
| Medications (e.g., amphotericin, foscarnet, piperacillin, toluene) | ||
*Just a note on GI potassium handling: Under normal physiologic conditions, fecal potassium excretion accounts for <10% of potassium excretion. Therefore, the gastrointestinal tract is not normally a significant mechanism for regulating potassium homeostasis.
Potassium is the major intracellular cation, and less than 2% of total body potassium is in the extracellular compartment. Of note, the vast majority of potassium (~70%) is found in our muscles (which will become relevant when discussing our patient). The figure below outlines the distribution of potassium in our body.
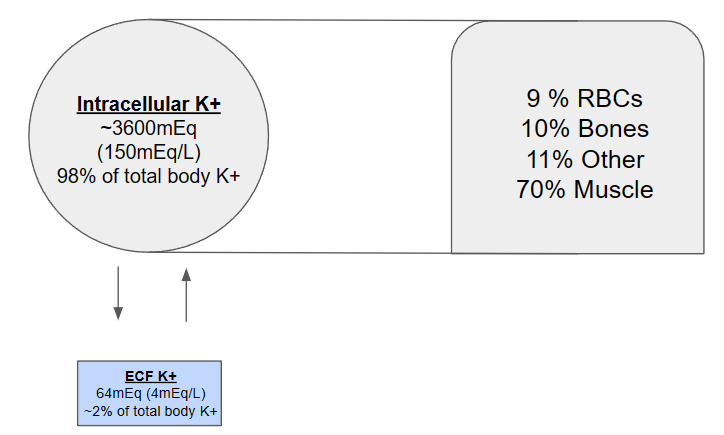
Percentages of distribution obtained from Comprehensive Clinical Nephrology, edited by Richard J Johnson, MD, 2024.
The major physiologic stimulus for inward potassium movement into cells include insulin and catecholamines by activating the insulin receptor and beta-2 adrenergic receptor, respectively. This results in increased Na-K ATPase activity causing sodium to move out of cells and potassium into cells. These transcellular shifts effectively “buffer” ECF potassium gains. In essence, hiding the ECF potassium in cells for a brief period of time. This was shown by Sterns et al by infusing potassium into anuric patients and measuring changes in plasma potassium. Under the assumption of a two compartment model, they demonstrated that only ~15% of potassium remained in plasma. The net potassium gains are then only transiently “buffered” by cells with the goal being eventual kidney excretion. While surges in catecholamines, perturbations in acid-base homeostasis, cellular lysis, or insulin deficiency cause cellular shifts that change the plasma potassium concentration, the kidney’s excretory ability maintains total body potassium balance.
Potassium’s Journey

Please sit back, relax, and join me on a not-so-lazy river as we follow potassium’s journey through the nephron and look into the causes of potassium loss at each stop.
A. Bowman’s capsule and the proximal convoluted tubule (PCT)
The first stop on our journey is Bowman’s capsule. Like other solutes, the potassium concentration of the filtrate within Bowman’s capsule is the same as that of plasma. As we enter the PCT, 65-70% of filtered potassium is passively reabsorbed through the paracellular route via solvent drag. The basolateral Na/K ATPase maintains a gradient that favors Na+ entry into the PCT cell. This movement in Na+ results in movement of H2O and other electrolytes-including potassium- from the tubular lumen into the PCT cell.
- Mechanisms of hypokalemia:
- Proximal RTA and proximal tubule dysfunction — see our Fanconi case written by Santhoshi Bavi
- Medications — Proximal tubule diuretics like SGLT2 inhibitors block sodium reuptake in the proximal tubule leading to increased sodium delivery to the distal tubule where it is exchanged for potassium (in clinical data, SGLT2 inhibitors reduce hyperkalemia but do not cause hypokalemia)
B. Loop of Henle (LH)
Only 10% of filtered potassium is left as we enter the LH. Thanks to the super-concentrated medullary interstitium, water is passively reabsorbed as tubular fluid enters the descending loop. After we turn the corner and begin our ascent, potassium is pulled from the tubular fluid thanks to the action of the Na/K/2Cl (NKCC) transporter located within the luminal membrane of the thick ascending limb loop of Henle.
- Mechanisms of hypokalemia
- Medications — Loop diuretics, a pharmacologic blockade of NKCC
- Bartters syndrome —a congenital defect that impairs NKCC
C. Distal convoluted tubule (DCT)
The next stop on our journey is the DCT. Potassium excretion is slightly favored at this point. Na flows down its concentration gradient into the cell through epithelial sodium channels (ENaC) resulting in a negative tubular lumen relative to the intracellular space. The negative potential promotes K movement out of the cell and into the lumen via ROMK channels. This is where magnesium comes into play. Intracellular magnesium binds to the cytosolic sites of ROMK, limiting K outflow. As a result, serum potassium concentration is inversely proportional (it’s the opposite) to intracellular magnesium concentration- as demonstrated by the figure below. Thus, hypomagnesemia can both cause and worsen pre-existent hypokalemia.
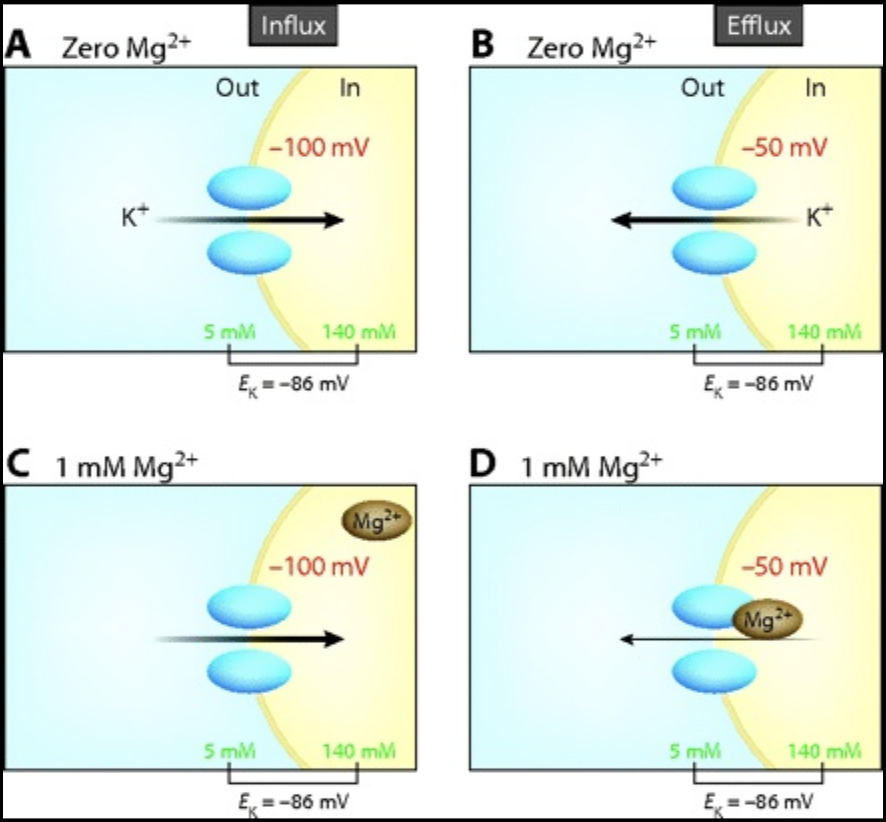
- Mechanisms of Hypokalemia
- Medications: Thiazide diuretics — pharmacological blockade of the sodium-chloride cotransporter (NCC). While NCCl is not directly responsible for K reabsorption, its blockade can lead to increased distal Na delivery feeding into the ENaC mechanism described above.
- Hypomagnesemia — as described above
- Gitelman syndrome — genetic defect in NCC.
D/E. Collecting duct (CD)
We have come to our last stop on our journey through the nephron. While the PCT is the workhorse for potassium reabsorption, the CD is the star of the show when it comes to potassium excretion. It is here that the rate of excretion can exceed the rate of reabsorption. The ROMK channel is the major passageway for K out of the cell and into the urinary space. There are also Maxi-K channels that are less physiologically important, but when mutations are present, can result in K wasting as seen in certain variants of Bartter’s syndrome (Type 2). The rate of K excretion is determined by: aldosterone, distal Na delivery, and tubular flow rate. Aldosterone activates the Na/K-ATPase, causing an increase in intracellular K, favoring K excretion. Aldosterone also increases Na reabsorption via ENaC, causing a negative tubular lumen drawing K into the urinary space. An increase in distal Na delivery will also lead to increased Na reabsorption via ENaC and greater K secretion. Increased tubular flow, as seen with high volume infusion of isotonic crystalloid solutions, results in a decreased luminal K concentration and thus promotes movement of intracellular K down its concentration into the lumen.
- Mechanisms of hypokalemia
- Hyperaldosteronism
- Distal/Type 1 RTA
- Bartter syndrome type 2
Workup of Hypokalemia
Now that we understand the life of potassium in our body, here are two helpful flowcharts for working up hypokalemia.
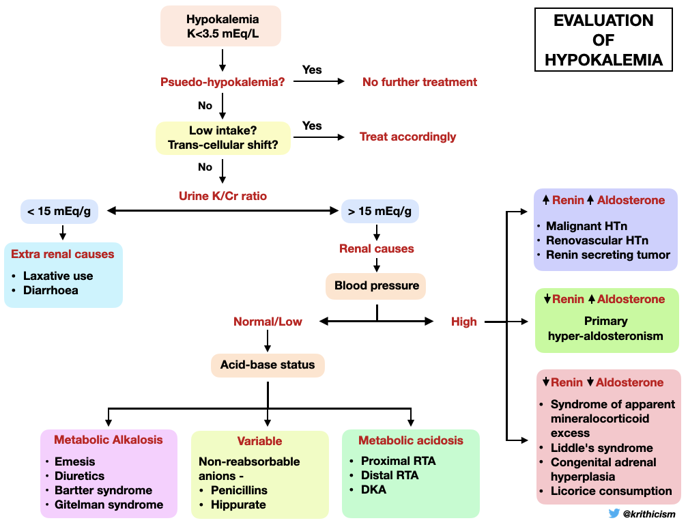
D. The Diagnosis: Thyrotoxic periodic paralysis
In summary, this was a 41-year-old man with chronic unexplained hypokalemia who presented with acute on chronic bilateral lower extremity weakness that progressed to total paralysis. His workup revealed severe hypokalemia in the setting of an undetectable TSH and elevated free T4 level. The combination of his demographics, clinical presentation with periodic episodes of paralysis, and serologies (undetectable TSH, elevated T4, elevated TSI, hypokalemia) is suggestive of thyrotoxic periodic paralysis (TPP).
TPP belongs to a family of channelopathies associated with hyperthyroidism. Grave’s disease is the most common underlying condition, but any other cause of elevated thyroid hormone, including exogenous use, can trigger it. In addition to hypokalemia, findings suggestive of TPP include:
- Elevated T4
- Low T3
- Mild hypophosphatemia
- Mild hypomagnesemia
- More common in East Asian ancestry individuals
- More common in men than women
- Often presents after the 2nd decade of life
Since all periodic paralysis clinically present similarly and treatments differ, distinctions between hypokalemic periodic paralysis (HPP) and thyrotoxic periodic paralysis (TPP) is important.
HPP is an autosomal dominant disorder affecting the dihydropyridine-sensitive calcium and sodium channels within skeletal muscles. It often occurs within the first and second decades, and is treated with carbonic anhydrase inhibitors and potassium sparing diuretics.
TPP, on the other hand, is thought to be related to increased activity of Na-K ATPase. Elevated thyroid hormone activates sympathetic tone, which further drives Na-K ATPase activity and drives potassium into cells. That is why other causes of increased sympathetic tone, such as stress and intense physical activity, can also trigger paralysis in these patients. Additionally, increases in sympathetic tone results in increases in the number and the sensitivity of beta-adrenergic receptors to catecholamines, which further drive intracellular potassium shifts. It is hypothesized that thyroxine is structurally similar to catecholamines and may exert effects on catecholamine receptors. Therefore, nonselective beta blockers are effective in treating hypokalemia associated with TPP as we will see later.
In addition to laboratory findings as above, testing for genetic susceptibility to TPP is difficult partly due to the small number of cases and absence of a familial pattern of inheritance. The known genes associated with HPP (CACN1AS and SCN4A) have mostly been negative in TPP patients. The KCNJ18 gene encodes potassium channel Kir2.6 found on skeletal muscle, and mutations have been found to be present in 33% of TPP patients resulting in a defect in muscle repolarization. The INVITAE panel will test for the majority of these mutations except for KCNJ18.
If you have not seen the videos of “the fainting goats” online, we highly recommend doing that. These myotonic goats have a defect in a chloride channel leading to a decrease in intracellular chloride, which results in potassium moving extracellularly to balance the ion gradient. However, potassium accumulates at skeletal muscle T-tubules and depolarizes myocytes leading to muscle stiffness and the subsequent “fainting goat”. See the excellent review on the ClC-1 chloride channels by Pederson et al for further detail.
On the flip side, in patients with TPP, thyrotoxicosis leads to a combination of factors that ultimately increases intracellular potassium including:
- Increasing the transcription and activity of the Na-K ATPase
- Raising catecholamine levels
- Inhibiting Kir (potassium leak) channels.
All the above effectively lowers extracellular potassium and causes hyperpolarization with resultant episodic paralysis.
The core tenets of treating TPP include potassium supplementation and correcting the underlying thyroid disease. Those treated with IV potassium tend to recover more quickly than those receiving oral potassium supplementation. However, remember that rebound hyperkalemia occurs in 40-60% of patients, with a higher prevalence in those with higher thyroxine levels and who require higher potassium supplementation. IV propranolol can also be considered in refractory patients by reversing excessive stimulation of sodium-potassium ATPase and subsequent intracellular drive of potassium. Avoiding high-carbohydrate meals and heavy exercise are important lifestyle modifications. In an observational study of 135 patients with TPP, the most commonly reported triggers were high-carbohydrate meals (12%), upper respiratory tract infection (8%), and strenuous exercise (7%). Ultimately, the main therapy is to restore euthyroidism with the assistance of our endocrinology colleagues.
Unfortunately, this patient developed anuric kidney failure after his cardiac arrest and remained dialysis dependent until discharge. Therefore we were unable to obtain urine studies to demonstrate renal wasting of potassium. Additionally, he was lost to follow up and we were unable to perform genetic testing on him. However, his history, symptoms, demographics, and thyroid studies supported the diagnosis of thyrotoxic periodic paralysis.
E. Take home points
- The differential diagnosis of hypokalemia includes potassium redistribution, renal losses, or non-renal losses
- Thyrotoxic periodic paralysis presents with episodic weakness with hypokalemia and laboratory findings consistent with hyperthyroidism
- Potassium supplementation and propranolol are treatments in the acute phase of thyrotoxic periodic paralysis, however, rebound hyperkalemia may occur
- Ultimately, treatment of the underlying thyroid disease is needed
Reviewed by: Santhoshi Bavi, Chi Chu, Sai Achi, Jefferson Triozzi, Raad Chowdhury, Joel Topf, Matthew A. Sparks, Margaret DeOliveira

