Welcome to the 39th case of the Skeleton Key Group, a team of nephrologists from around the world who build a periodic education package for the Renal Fellow Network.
Author: Saud Alsaleh (@Saud___Alsaleh)
A. The Stem:
A 42-year-old man with no prior medical history presented with a sudden, severe headache and altered mental status. An emergent evaluation revealed a subarachnoid hemorrhage with intraventricular extension. He required neurosurgical intervention, including placement of an external ventricular drain (EVD) and coil embolization of a ruptured cerebral aneurysm. The procedure was complicated by an intraoperative thrombus, and the hospital course was further complicated by accidental EVD dislodgement, leading to gram-negative meningitis treated with IV antibiotics.
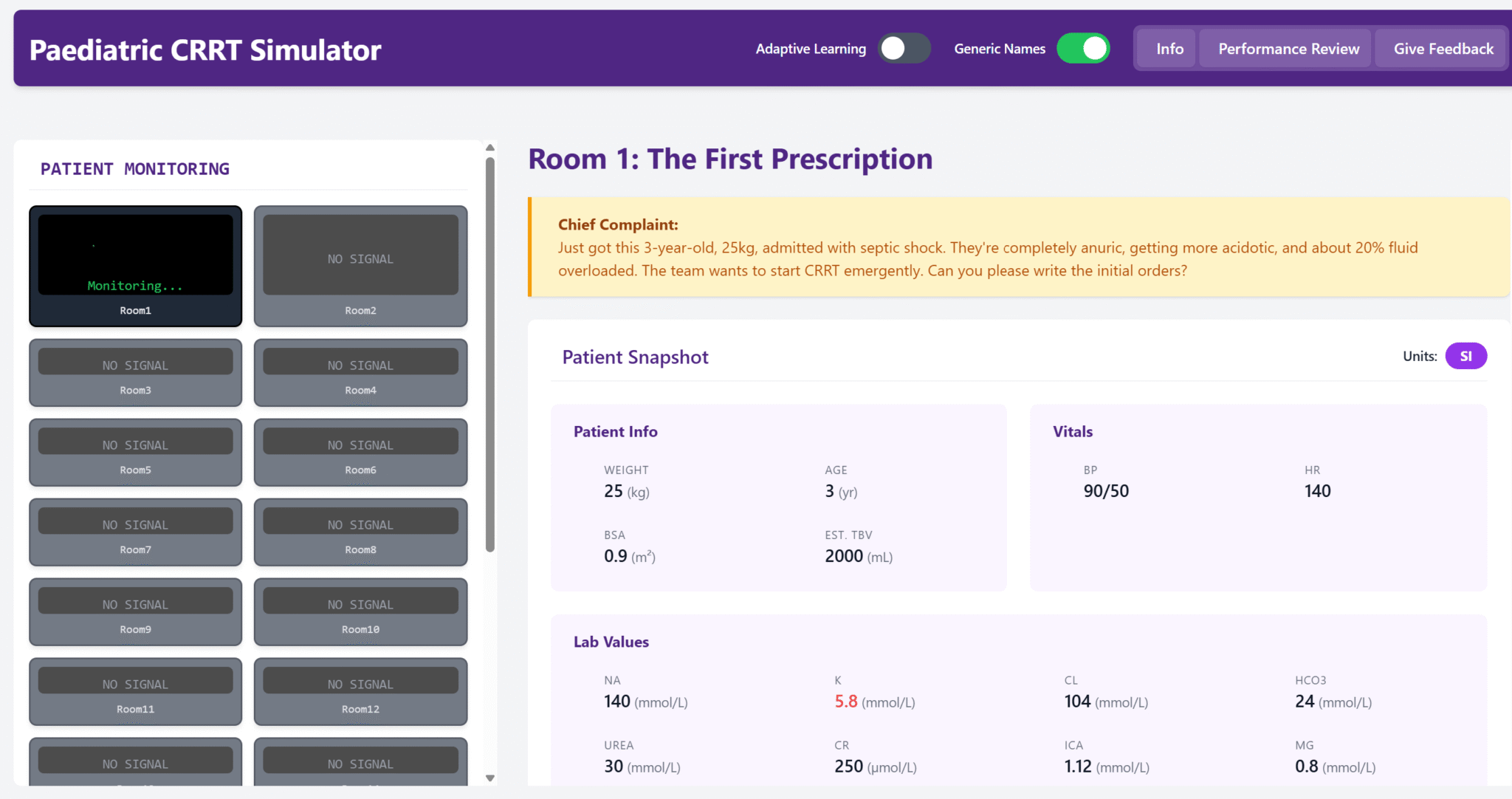
During the second week of hospitalization, the patient developed hyponatremia and polyuria. His serum sodium nadir was 125 mEq/L, accompanied by urine output of up to ~5-6 L/day. He developed increasing confusion and lethargy. Neurocritical care initially managed the hyponatremia with hypertonic (3%) saline boluses, oral salt tablets, fludrocortisone, and fluid restriction. Despite these interventions, his sodium fluctuated in the 125–132 mEq/L range, rebounding only transiently with 3% saline and then drifting down again. He endorsed lightheadedness on standing. Notably, an SSRI (citalopram) had also been started for mood during the hospital stay. The nephrology service was consulted for further evaluation and management.
Vital Signs:
- T: 97.5 F (36.4 C)
- BP: 125/95 mm Hg
- HR: 95 bpm
- RR: 12 per minute
Orthostatic Vital Signs:
BP 125/95 mm Hg supine, dropping to 99/83 mm Hg upon standing, with a compensatory tachycardia from 120 up to 130 bpm.
Physical exam:
- Gen: alert and oriented to person
- HEENT: dry oral mucous membranes
- Card: Elevated rate, regular rhythm, flat jugular veins
- Pulm: clear to auscultation bilaterally
- Abd: soft and nontender
- Ext: No peripheral edema
- Neuro: Grossly normal, no focal deficits
- Psych: Apathetic affect
B. The Labs
Serum osmolality: 270 mOsm/kg
Urine osmolality: 599 mOsm/kg
Urine sodium: 104 mEq/L
Urine uric acid: 41 mg/dL;
Fractional excretion of urate (FEurate): 13% (elevated above normal, >11–12%).
Serum uric acid: ranged 1.9–3.1 mg/dL on serial checks (low).
Urinalysis: Specific gravity 1.020, no protein, glucose, or ketones; no blood or infection markers.
Complete blood count: WBC 6.8 x10³/µL, Hemoglobin 10.3 g/dL, Platelets 474 x10³/µL (reactive thrombocytosis).
Thyroid function: TSH 4.5 µIU/mL (upper-normal), Free T4 0.8 ng/dL (normal).
Adrenal function: 8 AM cortisol 11.1 µg/dL (borderline low-normal).
Plasma renin activity: 9.3 pg/mL; Aldosterone: 3.9 ng/dL (Aldosterone-renin ratio ~0.4). (These renin-aldosterone levels were not frankly elevated, as might be expected in hypovolemia, but also not appropriately suppressed, more on this later.)
B-type natriuretic peptide (BNP): 26 pg/mL (normal).
C. The Work-Up
Approach to Hyponatremia Diagnosis
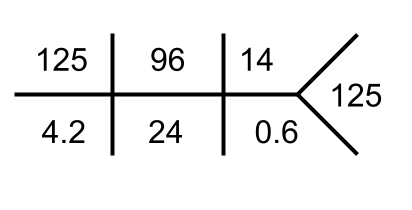
The initial workup of hyponatremia begins with measuring serum osmolality to determine if it is true hypoosmolar hyponatremia; the normal reference range for serum osmolality is 275–295 mOsm/kg. If hypoosmolar hyponatremia is confirmed, the next step is to assess urine osmolality, which provides insight into antidiuretic hormone (ADH) activity. High urine osmolality suggests increased ADH activity, leading to water retention. Additionally, measuring urine sodium concentration helps evaluate the renin-angiotensin-aldosterone system (RAAS) activity; a urine sodium level greater than 20 mEq/L may indicate suppressed RAAS activity such as in SIAD or renal salt wasting, while a level less than 20 mEq/L suggests increased RAAS activity such as in hypovolemia or edematous states like heart failure or cirrhosis.
By integrating these laboratory findings with the patient’s clinical status, we can classify hyponatremia based on volume status (hypovolemic, euvolemic, or hypervolemic) and identify the underlying cause to guide appropriate management
In this case, the patient’s labs showed hypotonic hyponatremia (Serum osm was 270 mOsm/kg) with inappropriately concentrated urine (high Uosm, 599 mOsm/kg) and high urinary sodium losses (104 mEq/L). Thyroid and cortisol levels ruled out hypothyroidism and Addison disease. The combination of low serum uric acid, high FEurate, and inappropriately high urine Na suggested SIAD or cerebral salt wasting (CSW) in the context of neurological injury.
Before diving in, let’s step back and walk through the systematic approach that led us to this differential.
Taking a Closer Look
1. Serum Osmolality – Confirm Hypotonic Hyponatremia: Hyponatremia can be hypotonic (true “water excess”), isotonic, or hypertonic. The first step is to measure serum osmolality. In our case, measured serum Osm ~270 mOsm/kg confirmed a hypotonic hyponatremia, as expected with his low [Na⁺]. (For reference, a serum Osm <275 mOsm/kg virtually always indicates true hypotonic hyponatremia. We should always rule out other scenarios. Iso-osmolar (pseudohyponatremia) can occur with severe hyperlipidemia or paraproteinemia, causing a lab artifact. This happens because widely used serum multianalyzers measure Na+ using indirect potentiometry and specimen dilution, which assumes that water constitutes 93% of plasma. Using direct potentiometry will avoid this dilution error. Furthermore, hyperosmolar hyponatremia occurs when an osmotically active solute (glucose, mannitol, glycerine, sorbitol, radiographic contrast agents, glycine, or intravenous immunoglobulin) accumulates and draws water out of cells diluting sodium (See SKG Case #8 by Jefferson Triozzi for more on this).
Our patient’s normal glucose and low serum osmolality ruled out those possibilities. In short, he had a true hypotonic hyponatremia: there was too much free water relative to solute in his body.
2. Urine Osmolality: Is ADH On or Off? In hypotonic hyponatremia, urine concentration tells us whether antidiuretic hormone (ADH) is in play. A maximally dilute urine (Uosm <100 mOsm/kg) indicates the kidneys are appropriately excreting free water (i.e. ADH is suppressed) as is seen in primary polydipsia or beer potomania. ADH secretion can be appropriate (e.g. due to true volume depletion or hyperosmolarity) or inappropriate (SIAD). Given his hypotonic state, osmoreceptor-driven ADH release (which normally responds to high osmolarity) should have been off, so we suspected non-osmotic triggers of ADH. Pain, nausea, CNS disturbance, and medications (like SSRIs) are known non-osmotic triggers, as is decreased perfusion (via baroreceptors). Thus, a high urine osm in this context narrowed our differential to causes of ADH-dependent hyponatremia – either hypovolemic hyponatremia or euvolemic SIAD (or a combination).
In our patient, Uosm was 599 mOsm/kg, which is very concentrated. This implies ADH is actively being secreted, causing the kidneys to retain water. In other words, some stimulus for ADH release was present despite his low sodium.
3. Urine Sodium and Volume Status: Is ADH Appropriate or Inappropriate?: Next, we assessed the urine sodium (UNa) in tandem with the patient’s volume status to differentiate appropriate vs. inappropriate ADH secretion. In true volume depletion, the body mounts a sodium-conserving effort via the renin-angiotensin-aldosterone system (RAAS); one would expect a low UNa (<20 mEq/L) as the kidneys avidly reabsorb salt to replenish volume. In SIAD (euvolemic hyponatremia), excess ADH causes water retention leading to mild volume expansion, suppression of RAAS, and thus a high UNa (>40 mEq/L) as the body excretes sodium to reach a new steady state (See SKG Case #24 by Sai Achi for more on SIAD).
Our patient presented a bit of a paradox: his exam suggested volume depletion (orthostatic, tachycardic, dry mucosa), yet his urine sodium was 104 mEq/L, which is quite high. A high UNa in the face of apparent hypovolemia raised suspicion for cerebral salt wasting, a scenario of renal salt loss due to intracranial pathology. Importantly, we had to consider that clinical volume assessment is not foolproof. The patient had received hypertonic saline and salt supplementation, which could influence urine Na, and his neurologic injury and supine status might mask some edema if present. However, the high BUN:Cr ratio (~23) was also consistent with a volume-contracted state. Thus, the combination of high ADH activity (high Uosm) with inappropriately high urine Na and clinical hypovolemia pointed to a diagnosis of Cerebral Salt Wasting (CSW) rather than simple SIAD.
Some Caveats
It’s worth noting a few caveats in interpreting urine sodium and osmolarity. Advanced kidney disease can impair the kidney’s ability to dilute or concentrate urine, limiting maximum dilution to higher than 50-100 mOsm/kg. Our patient had normal kidney function, so this was not a factor. Low solute intake (beer potomania or tea-and-toast diet) can keep urine dilute due to ADH being off and not enough solute to excrete. Again, not an issue here as he was receiving nutrition. Diuretic use can raise UNa, reducing its usefulness as a marker of RAAS activity. In SIAD, if a patient is given significant sodium (e.g. hypertonic saline or salt tabs), the UNa might not be as low as expected once the load is excreted. Thus, trend and clinical context matter. Initially, our patient’s UNa was high prior to starting hypertonic saline, NaCl tabs, and fludrocortisone, and then continued to be elevated. This supported persistent natriuresis.
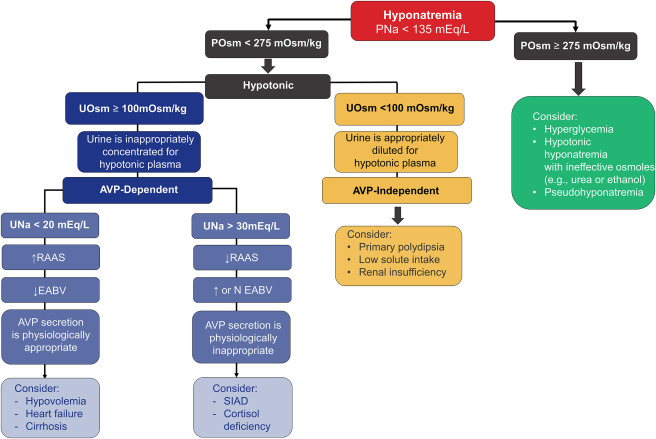
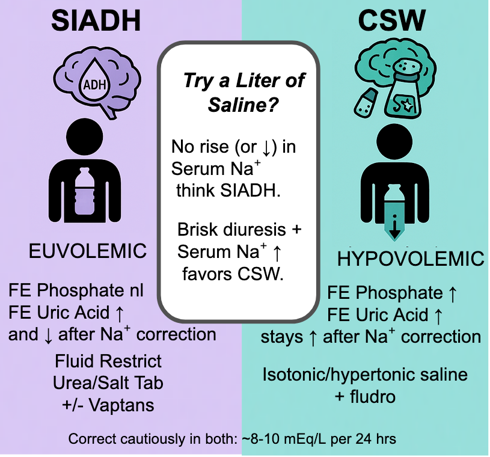
Differentiating SIAD from Cerebral Salt Wasting
Distinguishing CSW from SIAD is challenging because they share many lab features: both have hypotonic hyponatremia with high urine osm and high urine sodium. In both, serum uric acid tends to be low due to urate excretion in dilute plasma; FEurate is often >11% in SIAD and can also be elevated in CSW. Indeed, our patient’s FEurate was 13%, which is consistent with ADH-mediated urate clearance, seen in SIAD or CSW. Fractional excretion of phosphate is another possible option with promising results with limited evidence, initial studies have shown fractional excretion of phosphate is elevated in CSW (>20%) and normal in SIAD (<10%). Traditional teaching emphasizes volume status as the key differentiator. SIAD patients are clinically euvolemic (or even slightly hypervolemic), whereas CSW patients are hypovolemic by exam (signs of dehydration or volume contraction). Our patient clearly had signs of volume depletion. Additionally, his RAAS hormones were not elevated as one might expect in hypovolemia (perhaps due impaired sympathetic response in cerebral injury which is part of the pathophysiology of cerebral salt wasting).
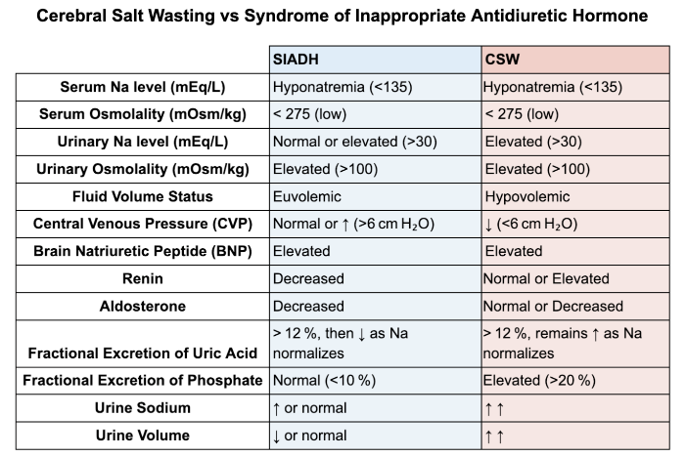
Table is compiled from information from review articles by Maesaka et al and Rudolph et al.
In practice, identifying CSW matters because the treatments diverge. SIAD is managed with fluid restriction, whereas CSW requires volume repletion. A therapeutic trial of isotonic saline can be revealing. if a hyponatremic patient improves with saline and demonstrates a brisk water diuresis once euvolemic, CSW is likely; if the sodium fails to rise or drops further with saline, SIAD is the main culprit. (Of course, in a patient with severe neurologic injury like subarachnoid hemorrhage, one would not withhold fluids for too long due to the risk of cerebral ischemia from hypovolemia. Our patient, in fact, did improve with saline administration, supporting a diagnosis of CSW).
Shoutout to NephMadness 2018: Hyponatremia Region for discussing SIADH vs CSW.
D. Diagnosis: Cerebral Salt Wasting (CSW)
This 42-year-old man with a subarachnoid hemorrhage and Serratia meningitis developed persistent hypotonic hyponatremia. Workup showed high urine sodium, elevated urine osmolality, and increased FEurate, consistent with cerebral salt wasting (CSW). Possible superimposed SIAD, likely due to neurological injury and SSRI use. He improved with isotonic saline, fludrocortisone, and sodium supplementation.
Now, let’s delve deeper into what CSW is and how we managed it.
Pathophysiology:
Cerebral salt wasting is a rare disorder of renal sodium loss triggered by intracranial disease, leading to hyponatremia and a contracted extracellular volume. Two major hypotheses explain CSW: (1) Sympathetic nervous system impairment: Neurologic injury (e.g. subarachnoid hemorrhage) can reduce sympathetic outflow and/or increase parasympathetic activity, resulting in decreased proximal tubular sodium (and water and uric acid) reabsorption, plus blunted renin and aldosterone release. The net effect is inappropriate natriuresis despite hypovolemia (and relatively low aldosterone levels, explaining why patients don’t retain sodium or excrete excess potassium). (2) Brain natriuretic peptide (BNP) release: Elevated intracranial pressure or direct brain injury can provoke release of BNP (or ANP) into the circulation. BNP promotes sodium and water excretion in the kidneys and suppresses renin secretion. Notably, some studies have found BNP elevations in subarachnoid hemorrhage patients with hyponatremia correlating with increased urine output and sodium loss, suggesting BNP as a mediator of CSW. (However, BNP can also rise in SIAD and in heart failure from the stress of CNS injury, so it’s not a perfect discriminator.) The exact mechanisms of CSW are still debated and some argue CSW might just be a misnomer for hypovolemic SIAD or an iatrogenic phenomenon. In our patient, BNP was normal, but he did have inappropriately low aldosterone for a volume-depleted state, supporting the idea of neurogenic RAAS dysfunction.
Clinical Manifestations:
CSW classically presents in patients with acute brain injury (aneurysmal subarachnoid hemorrhage being a prime example) within the first 1–2 weeks. Patients develop increasing urine output (polyuria) and natriuresis, leading to volume contraction and hyponatremia. Signs of volume depletion such as hypotension, tachycardia, low central venous pressure, hemoconcentration (elevated hematocrit), or pre-renal azotemia may be present. Our patient fit this pattern: about a week after his hemorrhage and neurosurgery, he became progressively hyponatremic with orthostasis and high urine output. This timing and presentation align with CSW, which typically emerges a few days post-neurologic insult and can last a few weeks. CSW has been reported with subarachnoid hemorrhage (being by far the most common culprit), traumatic brain injury, intracranial tumors, CNS infections (like meningitis/encephalitis), and after neurosurgery.
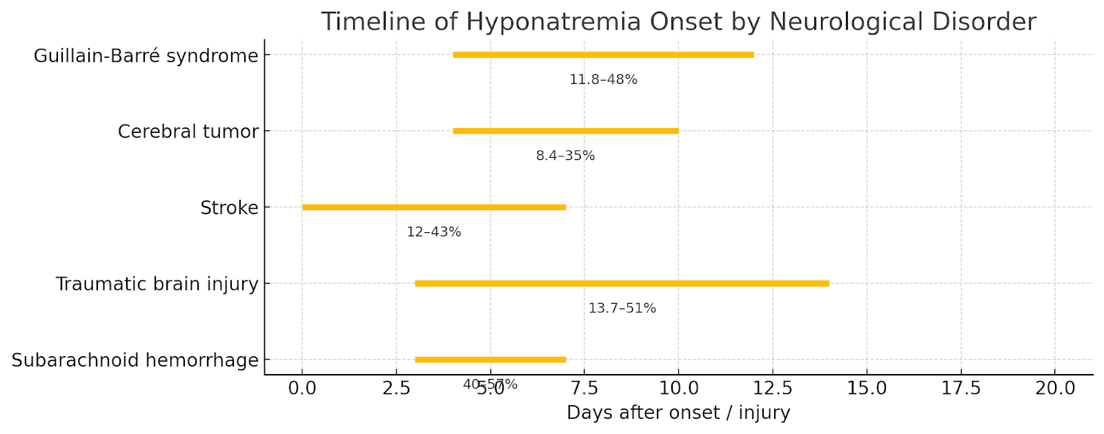
Figure 3 Hyponatremia timeline characteristics in different neurological disorders based on an observational review. This timeline was developed based on a table from Cui et al. Shows typically timeframes reported in the literature.
Diagnostic Work-up:
There is no single definitive test for CSW. It remains a diagnosis based on the constellation of labs and clinical findings. Key features include hypotonic hyponatremia, high urine osmolality (typically >300 mOsm/kg), inappropriately high urine sodium (often >40 mEq/L), low serum uric acid with high FEurate, and, importantly, evidence of volume depletion on exam or hemodynamics.
Our case checked all these boxes. It’s crucial to ensure no other cause of hyponatremia is present: in his evaluation we ruled out adrenal insufficiency (cortisol ~11, and he had no hyperkalemia or hypotension out of proportion) and hypothyroidism. SIAD was the main alternative diagnosis – and in fact, SIAD and CSW can co-exist, or CSW can evolve into SIAD as volume status changes. One suggested diagnostic maneuver, as mentioned, is volume resuscitation: in pure CSW, restoring volume should turn off the hypovolemic stimulus for ADH and result in a water diuresis and correction of sodium. In SIAD, giving volume tends to further dilute sodium. In practice, we gave our patient intravenous isotonic saline at 200ml/hr while closely monitoring sodium; his sodium improved gradually over 72 hours without signs of fluid overload, supporting CSW. Some experts also monitor changes in FEurate: in SIAD, once hyponatremia is corrected, the FEurate often normalizes (drops below 10-12%), whereas in CSW, FEurate may remain elevated until volume is fully restored. In our patient, FEurate stayed elevated during hyponatremia and we did not reassess it after correction, but the persistent natriuresis despite improvements in sodium suggested ongoing salt wasting. It’s worth noting that a comprehensive prospective study of subarachnoid hemorrhage patients by Hannon et al. found SIAD and hypovolemia is far more common than true CSW. In 100 patients with subarachnoid hemorrhage, about half developed hyponatremia, mostly from SIAD or corticosteroid deficiency, and none met strict criteria for CSW. This underscores that CSW is an uncommon (some say overestimated) diagnosis; one should be careful to meet all criteria (including clear volume depletion) before labeling a case “CSW.”
Follow-up:
The timeline of hyponatremia guides urgency of treatment. Acute hyponatremia (developing in <24–48 hours) water enters brain cells due to the hypoosmolar state, causing cellular swelling. In response, water moves from the brain parenchyma into the CSF and subsequently into systemic circulation to reduce ICP, brain cells expel extracellular ions (such as Na, K, Cl) to reduce intracellular osmolality, this helps reduce water influx and limit swelling, brain’s ability to reduce its volume through electrolyte loss is limited to approximately 18%.
In our patient, therapies for volume and sodium repletion (0.9% saline as infusion, salt tablets, fludrocortisone, plus liberalizing his fluid intake) gradually stabilized his sodium in the low-130s mmol/L. As his neurological condition improved and the meningitis cleared, his urine output normalized and sodium losses abated. After about 2–3 weeks, we were able to taper off the NaCl tablets and fludrocortisone. His serum sodium remained stable in the mid-130s with an unrestricted diet. CSW is usually transient, often resolving within 3–4 weeks as the acute brain injury phase passes. True to form, by the time of his rehab transfer, our patient’s hyponatremia had completely resolved and he no longer required salt supplementation.
E. Takeaway Messages
- Cerebral salt wasting is a hypovolemic hyponatremic syndrome seen in patients with acute neurological insults, characterized by excessive renal sodium loss and volume depletion.
- CSW is caused by dysregulated neural control of sodium homeostasis and involves impaired sympathetic tone, elevated natriuretic peptide levels (particularly BNP), and inappropriate renal sodium excretion.
- The diagnosis of cerebral salt wasting is suggested by the presence of hyponatremia with hypovolemia, inappropriately high urine sodium concentration (>50-100 mmol/L), and an elevated fractional excretion of urate (FEurate >11%), in the setting of hypovolemia and polyuria, typically in the setting of acute neurological injury.
- In differentiating cerebral salt wasting and SIAD, clinical Volume Status is Key: Hypovolemia suggests CSW (e.g., orthostatic hypotension, tachycardia), while euvolemic or mild hypervolemia indicates SIAD. A therapeutic trial of isotonic saline can help differentiate them.
- CSW requires volume and sodium repletion, whereas SIAD is managed with fluid restriction.

Editors: Jefferson L Triozzi, MD MSCI, Joel Topf, Chi Chu, Matthew A. Sparks MD, Srinath Yadlapalli, MD.