Welcome to the 24th case of the Skeleton Key Group, a team of 50-odd nephrology fellows who work together to build a monthly education package for the Renal Fellow Network. The cases are actual cases (without patient identifying information) that intrigued the treating fellow.
You’ve learned about hyponatremia in our 3rd case by Amy Yau, and 20th case by Bilal Sheik, but we always love a classic!
Written by: Sai Achi
Visual Abstract by: Priti Meena
A. The Stem
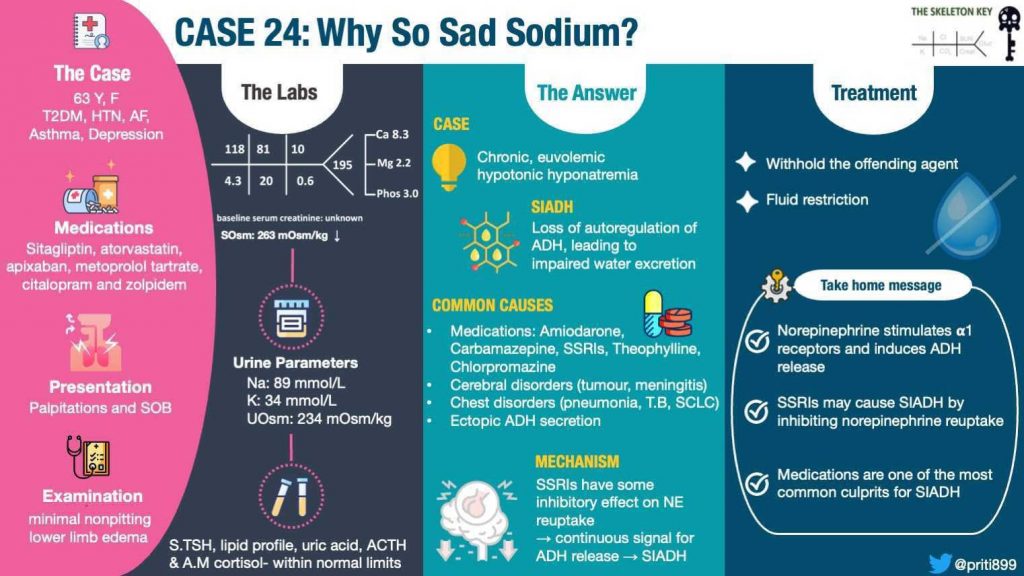
A 62-year-old woman with a history of type 2 diabetes mellitus, hypertension, asthma, atrial fibrillation (on apixaban), and depression presented with several hours of palpitations and shortness of breath.
She initially attributed the symptoms to asthma exacerbation and took her nebulizers without relief, prompting a visit to the emergency department. She is a former smoker who quit 21 years ago. She does not report any alcohol, recreational drug use, or family history of kidney disease. A couple of months ago, her primary care physician told her that her sodium was “on the lower side.” She also notes decreased appetite lately.
Her home medications include sitagliptin, atorvastatin, apixaban, metoprolol tartrate, citalopram, and zolpidem.
Physical Exam:
Vitals on arrival to the ED: temperature 98.1 F, heart rate 121 beats per minute, respiratory rate 20 breaths per minute, blood pressure 152/94, oxygen saturation 100% on room air
Physical exam: the patient is alert, and oriented to person, place, and time. She is tachycardic, with an irregularly irregular heart rhythm, has clear breath sounds without use of accessory muscles, no stridor, soft abdomen, and edema up to the mid calf with minimal indentation and immediate rebound upon pressing the lower extremity.
B. Labs
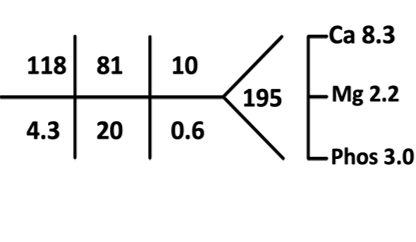
Serum osmolality: 263 mOsm/kg
AM cortisol: 9.65 mcg/dl (reference range: 4.0 mcg/dl-22.4 mcg/dl)
ACTH: normal (done simultaneously with AM cortisol)
Uric acid: 5.5 mg/dl
Total cholesterol: 142 mg/dl
Triglycerides: 114 mg/dl
HDL: 98 mg/dl
LDL: 22 mg/dl
TSH : 3.98
Urine studies:
Na: 89 mmol/L
K: 34 mmol/L
Urine output was 1000 ml/day
Urine osmolality 234 mOsm/kg
UA: pH 6.2; specific gravity 1.006 (reference range: 1.003-1.035); negative for protein, glucose, ketones, leukocyte esterase, nitrites, wbc, rbc, casts
CXR: clear lungs
Nephrology was consulted for hyponatremia.
C. Working Up the Case
Understanding the timeline of development of the hyponatremia is important, particularly for determining a safe rate of sodium correction. In our patient, the hyponatremia is chronic (usually defined as >48 hours). When we don’t know when exactly the hyponatremia started, we also assume that it is chronic because you do not want to even risk guessing and overcorrecting to quickly which can have permanent neurological sequelae. After reviewing the algorithm below, let’s follow our patient’s labs and see what type of hyponatremia we have.
The differentials and algorithms for hyponatremia include those in the following graphic.
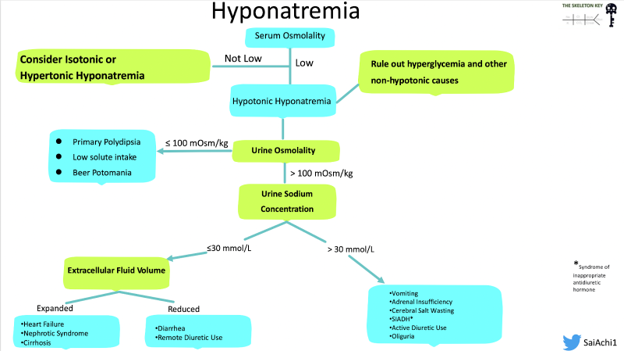
Her serum osmolality is 263, which is low. This allows us to say that the patient has true hypotonic hyponatremia, rather than non-hypotonic (i.e., isotonic or hypertonic) hyponatremia. There were no clues to suggest hyperosmolality, such as iodinated contrast use, history of receiving mannitol, or any isotonic causes such as hyperlipidemia at play here.
For more information on non-hypoosmolar hyponatremia, please check out this SKG case from Jefferson Triozzi.
Now that we have established that the patient has hypotonic hyponatremia, the next step is to look at the urine osmolality to gauge whether or not the kidneys are diluting urine (excreting free water) appropriately, which would be the expected response to plasma hypotonicity. This tells us whether or not the hyponatremia is ADH dependent or ADH independent. As the function of ADH is to retain water, producing concentrated urine, a high urine osmolality would suggest an ADH dependent process, while low urine osmolality would suggest an ADH independent process. Then after using the information from the urine osmolality we come to determining volume status. Since relying on the physical exam alone is challenging, we can use the urine sodium to further aid us as we work through the algorithm (Figure 2). After we have the high urine osmolality (Urine Osmolality: 234 mOsm/kg) confirming an ADH-dependent process, this figure shows how urine sodium can help break down the volume status aspect of the hyponatremia differential, although we should also note that it is not always black and white: there may be times when a patient may have multiple drivers of hyponatremia.
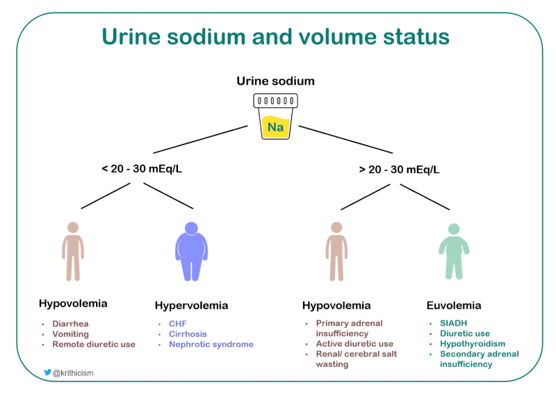
So far we have a patient who has true hypotonic hyponatremia with a urine sodium of 89 mmol/L. The high urine sodium indicates that the kidneys are not avidly retaining sodium, suggesting against states of effective arterial volume depletion. So this means that either the patient is not hypovolemic or is hypovolemic with an impairment of sodium reabsorption which is seen in active diuretic use, mineralocorticoid deficiency, or cerebral/renal salt wasting syndromes. In our patient, there were no clinical signs of hypovolemia. Additionally, she had no history of diuretic or chronic steroid use, with no history of unexplained hypoglycemia or hypotension, arguing against primary adrenal insufficiency.
What could then be causing the patient to have this picture of hyponatremia if she is not hypovolemic? Her TSH was normal, excluding hypothyroidism. She had a normal random AM cortisol and ACTH was normal. On our physical exam, we initially had signs of edema and looking through her previous records–we couldn’t find any evidence of heart failure, liver disease, or proteinuria to suggest nephrotic syndrome. Upon further history, she stated that she works in a field where she is constantly on her feet for prolonged amounts of time and had been advised to elevate her legs, which improves her edema. This further supports the limitation of relying on the physical exam alone and the need to combine all of the urine and serum studies when approaching hyponatremia.
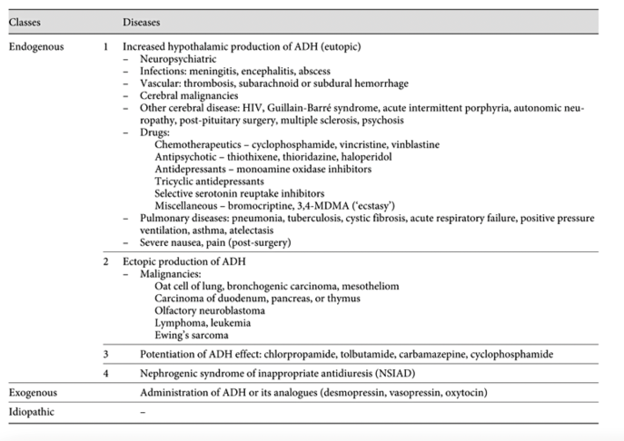
So we are left with SIADH. There are many causes of SIADH, a few of which are highlighted in Figure 3 below. Medications are one of the most common culprits. Upon a detailed review of medication history, citalopram was identified as the most likely cause of her hyponatremia and SIADH.
How do selective serotonin reuptake inhibitors (SSRIs) lead to SIADH?
In SIADH there is excess, unregulated ADH secretion leading to pathologic retention of water.
SSRI-induced SIADH can occur via three possible mechanisms:
- Excess ADH is secreted due to the effects of serotonin on 5HT2 and 5HT1c receptors
- Increased renal response to ADH
- Reset osmostat
ADH is secreted from the posterior lobe of the pituitary gland and is thought to be affected by factors such as angiotensin II, acetylcholine, endorphin and norepinephrine levels. In physiological situations, norepinephrine stimulates the alpha 1 adrenergic receptor to induce the release of ADH. Degradation of norepinephrine stops the release of ADH. A proposed mechanism is that while SSRIs inhibit the serotonin reuptake, they also inhibit norepinephrine reuptake, allowing continuous propagation of ADH release thereby leading to SIADH.
The following table highlights the risk of hyponatremia with the classes of antidepressants. Because the extent of the selectivity of dopamine, serotonin, and norepinephrine reuptake is different, the risk of developing hyponatremia via the SIADH mechanism differs by drug as well.
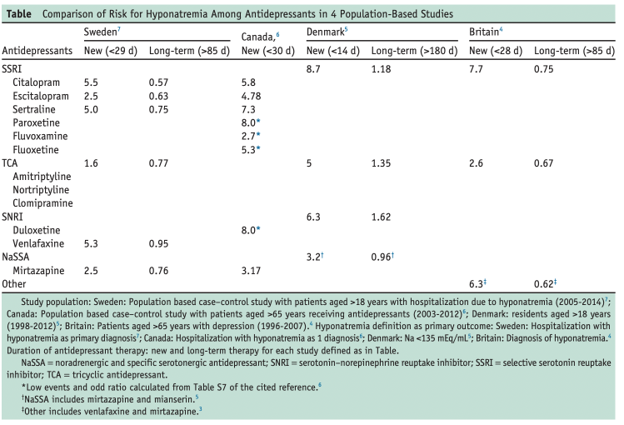
The table above relays the idea that across different countries, all SSRIs are associated with greater risk of hyponatremia overall in comparison to other classes of antidepressants. Citalopram, in particular, has a greater risk of new-onset hyponatremia compared to other SSRIs.
D . Final Diagnosis
Severe, asymptomatic, chronic, euvolemic, hypotonic hyponatremia secondary to SIADH in setting of citalopram use
E. Management
We restricted the patient’s fluids to 1L a day and the sodium increased initially by 4 mEq/L over the first 24 hours and continued to rise thereafter. Eventually the sodium improved to 136 mEq/L over 4 days.
Citalopram was discontinued and changed to mirtazapine with close follow-up with her psychiatrist and nephrologist at the outpatient clinic. Although there is a slight risk of hyponatremia with mirtazapine, she tolerated the medication well, with no notable decrease in her sodium since her last follow up.
F. Take Home Points
- In normal physiologic situations, norepinephrine stimulates alpha-1 adrenergic receptors and induces ADH release.
- SSRIs may cause SIADH by inhibiting norepinephrine reuptake allowing a continuous signal for ADH release.
- Medications are one of the most common culprits of SIADH. Prompt recognition is necessary to avoid adverse effects.
Reviewed and edited by Joel Topf, Dominique Tomacruz, Chi Chu, Alejandro Meraz, Nasim Wiegley, Matthew A. Sparks, Bilal Sheikh


I think I can answer Naveen and Kelly’s question about urine Osm – typically it is higher than serum Osm in SIADH but it is not necessarily as it is dependent on solute and water intake for the individual.
Say this woman takes in 400mOsm solute daily and drinks (minus insensible losses) 2L water. She is hypotonic, so her urine should be maximally diluted which for her is 234mOsm/L.
With that solute load she will only be able to produce 400/234=1.7L of urine per day. The extra 300cc would be retained as free water and worsen the hyponatremia.
This is also the rational for restricting water intake or increasing solute intake (salt tabs or urea) as a treatment for SIADH.
Hope that helps
Why did she have SOB? Was she congested with all the fluid retention?
Some gentle feedback:
Please help me understand Naveen’s question above. shouldn’t there be evidence of higher urine osmol than serum if ADH is present as is the assumption of SIADH?
The sitagliptan is known to cause CHF, in a patient with SOB and Le edema and known A fib, no role of evaluation for CHF here?
There are a lot of implications about discontinuing SSRI’s. I think we should not be cavalier to suggest this
Additionally, what happens when a patient has ATN? We cant use urine Na in that setting….
U Osm 234 baffles me a bit – “typically” would you not expect somewhat or a lot higher U Osm in SIADH ?
Calculated urine osmolality without including urea came as 246 (89+ 34)*2..How can measured urine osm be 234 most..
I would expect a role for low protein intake and so low urine urea (high protein intake would allow for lesser NA wastage in urine as the targeted urine osm that is set by ADH level can be reached easier by higher urea contribution ).. But even with zero protein intake, the endogenous catabolic process would still result in significant urine urea concentration
Great! Love me a hyponatremia. One point is that normal AM cortisol and ACTH does not rule out adrenal insufficiency unless in the setting of shock. It’s unlikely given that BP however to rule out adrenal insufficiency you should do a stimulation test.