I’ve always been skeptical about ACE inhibitors as a cause of hyponatremia, despite there being almost 20 case reports of this complication in the literature. Given the fact that so many patients I see are taking an ACE inhibitor, I’ve always felt I’d have encountered this problem more often if it were real. Anyway, I’ve now seen 3 convincing cases and have had to reconsider my position. The following is one such case, and is interesting as it suggests a possible mechanism:
Case: An elderly man with dementia and type 2 DM was admitted following a protracted diarrheal illness associated with poor oral intake. Clinically, he appeared volume depleted, although his blood pressure was normal. He was hyponatremic, with a high urinary sodium and high Uosm (numbers below). The primary team had given him a lot of saline (~6L over 48 hours), which aggravated his hyponatremia, but he continued to appear volume depleted. Urine sodium remained high (65-80), as did his urine output. Despite this Addisonian-type picture, the cort-stim test was normal; renin and aldo were pending.
We were called 3 days into the admission, and decided to stop his lisinopril and continue with normal saline. The results were pretty dramatic, as you can see from the graph below. He immediately became sodium avid (Una fell to less than10 meq/L) and had a water diuresis, correcting his plasma sodium to 132 meq/L over the next few days.
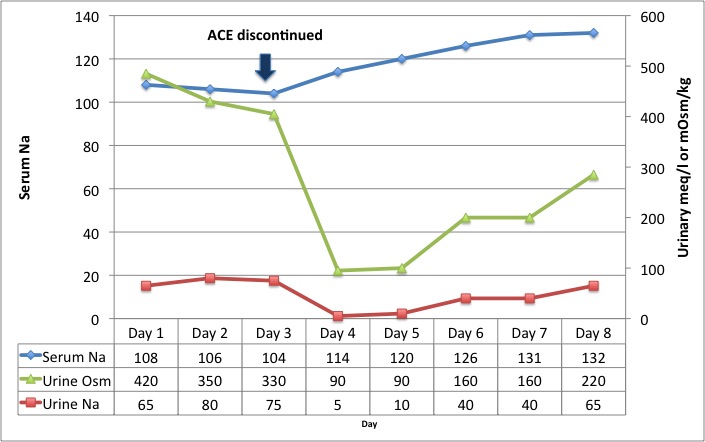
It seems to me that lisinopril was preventing a normal physiological recovery from volume depletion i.e. mineralocorticoid production. Once the ACE was stopped, he became salt-avid under the influence of aldosterone, allowing him to correct the volume depletion and switch off the physiological stimulus to ADH production (hence the water diuresis). This was not SIADH – his ADH axis was functioning normally, as evidenced by his ability to produce dilute urine and correct his plasma Na once the ACE was stopped.
The other 2 cases of ACE-associated hyponatremia I have seen have been remarkably similar to this, where severe volume depletion developed in a patient on chronic ACEi therapy. As such, in my limited experience, the presentation differs from other types of drug-induced hyponatremia, where the sodium falls shortly after the introduction of a new agent, say an SSRI or a thiazide. This insidious mode of presentation may explain why ACE-induced hyponatremia may not be on most people’s radar. Another reason for this may be that ACE-inhibitors can also paradoxically help correct hyponatremia in CHF patients, by improving cardiac output. I’m very interested to hear if anyone else has seen a similar case, as this is a controversial topic (as I’m sure the comment bar will attest!).
Hello,
I am a patient I have Fabry Disease. My Nephrologist is treating me with an ACE inhibitor lisinopril as well. My blood sodium is already on the low end of the numbers usually 136-138. It was 138 when started it went to 136. Now on it 3 months and it is 134 (I know this is not that low but want to make sure that it will not get worse) I was taking a look at my labs to see how im doing with the results of the meds and my alb/creat ratio. Seems like my labs are getting better besides the sodium which gets lower each time. Should I ask my Nephrologist about this or just keep an eye on my labs and see if there is changes?
I went down hard with hyponatremia in 2009 after 28 miles of running in rather warm tempaturs. I was 29 years old then. I was in a coma for 8 hours, they figured out I needed the saltier saline and I snapped out of it. Scary stuff, I was told it's because of the SSRI's I take. I still run Ultra's, my salt intake is 4 to 5 times higher than a normal athlete I guess because of the SSRIs. Tryed coming off the drug. Horrible experiance. I can tell now that I have to stop, let my body hydrate itself then keep going. Feels like my kidneys just shut down, then when I stop the flood gates come open. It's always a guessing game with how much salt to intake. I'm not a doctor if you couldn't tell by my writing. Anybody have the same thing going on?
I don't know if he has reset osmostat at all. If he did then, you would expect ADH production and thus more concentrated urine of day 8. His urine osm was 220, which is still not isoosmotic.
interesting too that his sodium didn't finally normalize, suggesting reset osmostat at baseline?
Serum K was 5.3 meq/l – no urine K results/TTKG unfortunately. Will need to dig out renin aldo results when I'm next in-house, they didn't come back during the time I was involved with this case.
I'm guessing that the primary stimulus to ADH release was volume contraction – as this improved with Na avidity, ADH activity fell and the Uosm dropped.
Thanks for interest, and don't forget to vote!
Sodium avidity following discontinuation of ACEI is understandable but how do you explain drop in the urine osmolality to <100 mosm?
Did you ever get the aldosterone and renin levels results?. secondly, was this patient hyperkalemic and do you have results of urinary potassium?
i had seen a patient recently who had unexplained hyponatremia, we just couldnt figure out why..now i remember he was on ACEI, and may be I should have discontinued ACEI and rechecked urine lytes and urine osmolality, next time will keep it in mind