 A 62-year-old man with CKD stage 3 secondary to presumed hypertension presented with new-onset hypercalcemia. PTH level was low at 11 and abdominal imaging revealed a right renal mass that later was confirmed to be a renal cell carcinoma.
A 62-year-old man with CKD stage 3 secondary to presumed hypertension presented with new-onset hypercalcemia. PTH level was low at 11 and abdominal imaging revealed a right renal mass that later was confirmed to be a renal cell carcinoma.
The most common cause of hypercalcemia of malignancy is secretion of PTH-related peptide, followed by increased bone resorption due to bone mets and increased 1-alpha hydroxilase activity by the tumor (similar to granulomatous disease).
His workup was remarkable for iCa 6.3, total Calcium 11.5, PO4 4.6, ACE level 36 and alk phos 109. Negative SPEP/UPEP, no elevation of 1,25 vitamin D and a mildly elevated PTHrP 49 pg/mL (14-27). One interesting aspect of PTHrP is that the most common assay measures the carboxy-terminal fragment, which is usually removed by the kidney and might be abnormality elevated in renal insufficiency. On our patient, his hypercalcemia (~11-13) was associated with worsening renal function and creatinine peaked at 4.3 mg/dl. Despite hydration and ibandronate 6mg IV, his hypercalcemia persisted at ~11-12 mg/dL (72 hours afterwards).
For further workup, another assay was performed with the amino-terminal fragment of PTHrP and revealed a low value (0.8). Lastly, there was no evidence of metastasis on bone scans. So what was the cause of the hypercalcemia in our patient and how can we get it under control?
Solid tumor cells are believed to secrete other bone-resorbing factors that mediate hypercalcemia through increased production of prostaglandins E (PGE). PGE is a potent stimulator of bone reabsorption in vitro and there is both animal and human data suggesting that PGE is an important contributor of hypercalcemia in some malignancies.
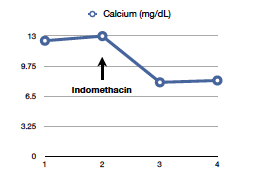
The most interesting aspect of it is that treatment involves administration of NSAIDS, which can block the production of PGE and resolves hypercalcemia. The original paper dates back to 1975 at the NEJM and there has been not many reports after that. Nonetheless, the team taking care of the patient started him on indomethacin 25mg q8hours as a last attempt to control his calcium before removal of the renal mass was performed. The results were astonishing as you can see in the graph above.
So despite all the negative effects of NSAIDs in the kidney, we have now a legitimate use in certain patients that have hypercalcemia 2/2 malignancy. Few questions still remain: how common is PGE-mediated hypercalcemia? Should we be checking for PGE levels as part of the workup? It seems that in patients that are not being considered for surgical resection and that did not respond to biphosphonates, a trial of NSAIDS could be warranted. Especially since the response is so quick!
Please share your experience as well since this was truly the first time I heard of this correlation. Kudos to the team taking care of this patient (led by Dr Mount) and Omar’s grand rounds’ presentation.
(Patient details modified to preserve patient identity)


Thanks Kenar for the questions. I was not clear on the days elapsed on the case… When indomethacin was given. more than 10 days had passed and 2 doses of biphosphonates had been administered with no response. Calcium remained elevated around 11-13. Because the response was so instantaneous with the indomethacin, it does seem it was the major player. IL-6 was not checked on this case.
Two things
Why don't you feel that the bisphosphonates kicked in around that time? usually they take some time to work and perhaps might have had some effect.
IL-6 has been associated with hypercalcemia in some cancers as well, was that checked?
Thanks Francesco for your comments. I do believe that malignancies could potentially have various mechanisms of hypercalcemia induction. Nonetheless, the abrupt response to NSAIDs is striking suggesting that PGE production was likely the dominant factor on this case. This is the first time I heard about the amino/carboxy terminals of PTHrP assays so I have to probably read more about it to find if there is any published work out there comparing both assays in patients with renal dysfunction.
Are you sure that the difference between amino-terminal and carbossi-terminal PTHrP is of clinical value? Why do you consider PGE production only as an alternative (not complementary)cause of hypercalcemia?