 I
Isaw a 20 year old African American male in clinic with nephrotic range
proteinuria, reduced GFR and no signs or symptoms of a systemic disease. His
mother developed ESRD at age 36 and his brother, aged 26, has CKD Stage
4. A full laboratory workup including complement proteins and ANA was
unremarkable. Renal biopsy demonstrated an FSGS pattern of injury with a full
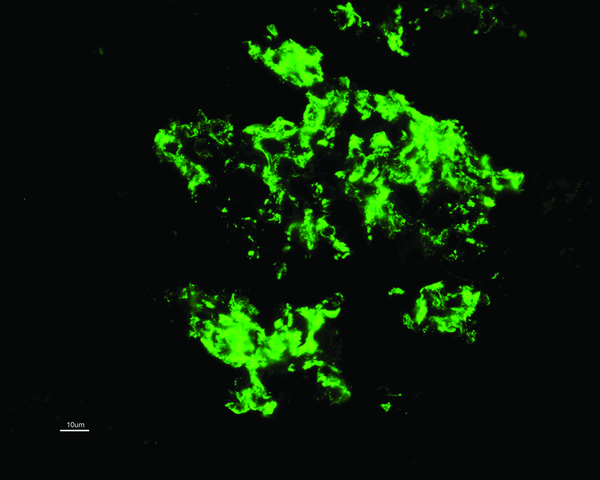
house picture on immunofluorescence but with dominant C1q staining. A diagnosis
of C1q Nephropathy (C1QN) was made.
Jennette et al described C1QN in 1985 in 15
patients without evidence of lupus who had dominant mesangial C1q deposition,
often with C3 and Ig’s also. The mean age at presentation was almost 18 years,
with a similar male: female ratio and average proteinuria of 7.5g/day.
Another series from Columbia University included 19 patients who were
predominantly African American (73%), young and female (73%) with nephrotic
range proteinuria. Renal outcomes across the series’ (including this European series) are mixed but would
suggest immunosuppressive treatment directed against the primary pathology
should be considered.
C1QN
may be considered an independent pathological entity, although this has been
controversial, with some classing it as a subtype of FSGS or minimal change
disease (MCD). The prevalence varies (0.2-16%) but is more common in children. The
predominant light microscopic appearance may be MCD, FSGS or a proliferative glomerulonephritis.
While C1q deposition (predominantly in the mesangium; see image) is dominant or co-dominant
by definition, a full house pattern of IgM, IgG, IgA, C1q and C3 may be
observed, more commonly with a proliferative glomerulonephritis. As for
treatment, the case series are small and largely uninformative. Some patients
appear to remit with or without immunosuppressive treatment whereas others have
a rapid course towards ESRD. Patients with an FSGS pattern may do worse than
other types. For what it’s worth, my patient had no response to 6 months of
steroids and mycophenolate and a plan for cyclosporine was made. The fact he
likely has a genetic element to his disease adds complexity. Some would argue
that immunosuppressive therapy in these cases is not likely to be of benefit
but we see from our hereditary FSGS cohort that this is not necessarily the case.
Cyclosporine and Rituximab in particular appear to have a directly beneficial
effect on podocyte function (also see case report of Rituximab treatment in
C1QN).
may be considered an independent pathological entity, although this has been
controversial, with some classing it as a subtype of FSGS or minimal change
disease (MCD). The prevalence varies (0.2-16%) but is more common in children. The
predominant light microscopic appearance may be MCD, FSGS or a proliferative glomerulonephritis.
While C1q deposition (predominantly in the mesangium; see image) is dominant or co-dominant
by definition, a full house pattern of IgM, IgG, IgA, C1q and C3 may be
observed, more commonly with a proliferative glomerulonephritis. As for
treatment, the case series are small and largely uninformative. Some patients
appear to remit with or without immunosuppressive treatment whereas others have
a rapid course towards ESRD. Patients with an FSGS pattern may do worse than
other types. For what it’s worth, my patient had no response to 6 months of
steroids and mycophenolate and a plan for cyclosporine was made. The fact he
likely has a genetic element to his disease adds complexity. Some would argue
that immunosuppressive therapy in these cases is not likely to be of benefit
but we see from our hereditary FSGS cohort that this is not necessarily the case.
Cyclosporine and Rituximab in particular appear to have a directly beneficial
effect on podocyte function (also see case report of Rituximab treatment in
C1QN).
As
for the pathogenesis and significance of the C1q deposits, we have much to
learn. Are the C1q deposits pathogenic or merely markers of injury? We do know that C1q binds to the basement
membrane via laminin and C1q receptors enhance binding of immune complexes to mesangial
cells where it combines with other complement proteins to form C1 protease.
This in theory allows activation of the classical complement cascade. Despite
this, serum C4 levels are generally normal and I can find no reports of complement
directed therapy (Eculizumab or C1-Inhibitors) in C1QN.
for the pathogenesis and significance of the C1q deposits, we have much to
learn. Are the C1q deposits pathogenic or merely markers of injury? We do know that C1q binds to the basement
membrane via laminin and C1q receptors enhance binding of immune complexes to mesangial
cells where it combines with other complement proteins to form C1 protease.
This in theory allows activation of the classical complement cascade. Despite
this, serum C4 levels are generally normal and I can find no reports of complement
directed therapy (Eculizumab or C1-Inhibitors) in C1QN.
Bottom
line: C1QN is likely an under recognized entity which can have varying clinical
and histological presentations. Renal outcomes are also variable and it may
have a different prognosis compared to traditional MCD (& FSGS?). One to keep in mind for the boards.
line: C1QN is likely an under recognized entity which can have varying clinical
and histological presentations. Renal outcomes are also variable and it may
have a different prognosis compared to traditional MCD (& FSGS?). One to keep in mind for the boards.
