Introduction
Fibroblast growth factor-23 (FGF23) is an osteocyte derived hormone that regulates serum phosphorus levels and vitamin D synthesis; inducing a negative phosphorus balance. Serum FGF23 levels increase early in chronic kidney disease (CKD). Furthermore, such elevations precede rises in serum phosphorus concentrations or hyperparathyroidism, suggesting a role beyond bone mineral regulation.
FGF23 has also emerged as a marker for cardiovascular complications, mortality and poor prognosis in patients with CKD. Interestingly, high FGF23 levels are detected early in the onset of acute kidney injury (AKI) as well, leading to the notion that it could be involved in its pathophysiology. This post aims to review classic and non-classic effects of FGF23 and its relevance in AKI/CKD and adverse clinical outcomes.
FGF23: biological role, classical/non-classic effects.
FGF23 is mainly secreted by osteocytes in response to hyperphosphatemia, high serum PTH, calcitriol, iron deficiency, and inflammation. One of the most important post-translational modifications is the cleavage of FGF23 into its biologically active form: intact-FGF23 (iFGF23). Byproducts such as C-terminal FGF23 (cFGF23) and N-terminal FGF23 are also released to the circulation as inactive peptides. iFGF23 exerts its effects through FGF23 receptors (FGFRs) inducing different cellular processes in classic target organs. The later, usually express klotho; a co-receptor that increases the binding affinity of FGF23 for FGFRs. For example, in the kidney, iFGF23 binds FGF23 receptor 1 (FGFR1) and its cofactor klotho to down-regulate the expression of 1-α hydroxylase and up-regulate 24-α hydroxylase; thereby limiting the concentrations of circulating calcitriol. In addition, it down-regulates the activity of the Na-phosphorus co-transporters; reducing phosphorus reabsorption.
Recently, it has been shown that FGF23 can also target cells that lack klotho and activate alternative downstream signaling pathways (“non-classic”). It is hypothesized that klotho-independent signaling mechanisms are activated in the presence of high FGF23 concentrations; ultimately leading to a myriad of pathologic cellular changes. For instance, one of the non-classic effects of FGF23 is mediated by FGFR4 and PLCγ/calcineurin/NFAT signaling pathway, which induces left ventricular hypertrophy and fibrosis in animal models. Reduced clearance by the kidney, increased extra-osseous production of FGF23 (e.g. spleen, kidney, and bone marrow) and inflammatory mediators are suspected to contribute to large serum concentrations of FGF23 in AKI/CKD (Figure).
FGF23, inflammation, and fibrosis
FGF23 can induce the expression of cytokines such as C-reactive protein and interleukin-6 in cultured hepatocytes. Such activation was elicited via FGFR4/PLCγ/calcineurin/NFAT signaling pathway in a 5/6 nephrectomy rat model of CKD. Furthermore, in a model of non-infectious (‘sterile’) inflammation using thioglycolate, Han X et al. showed that macrophages upregulate the synthesis of TNF-α mRNA through the FGFR and ERK activation. Interestingly, these macrophages were found to synthesize FGF23 as well, which could suggest a feed-forward effect and perpetuation of inflammation.
Smith et al. demonstrated that renal tubule-derived FGF23 can activate myofibroblasts and induce fibrosis via transforming growth factor β- related pathways in a model of acute interstitial nephritis. However, stimulation of fibroblasts derived from healthy kidneys did not elicit pro-fibrotic changes. Thus, this study suggested that FGF23 may induce pro-fibrotic signals in injury-primed fibroblasts, which can ultimately affect renal function beyond the original insult. Furthermore, treatment of injured kidney fibroblasts with soluble Klotho causes a switch from calcineurin/NFAT to Ras/MAPK signaling in isolated injured kidney fibroblasts; thereby decreasing fibrosis.
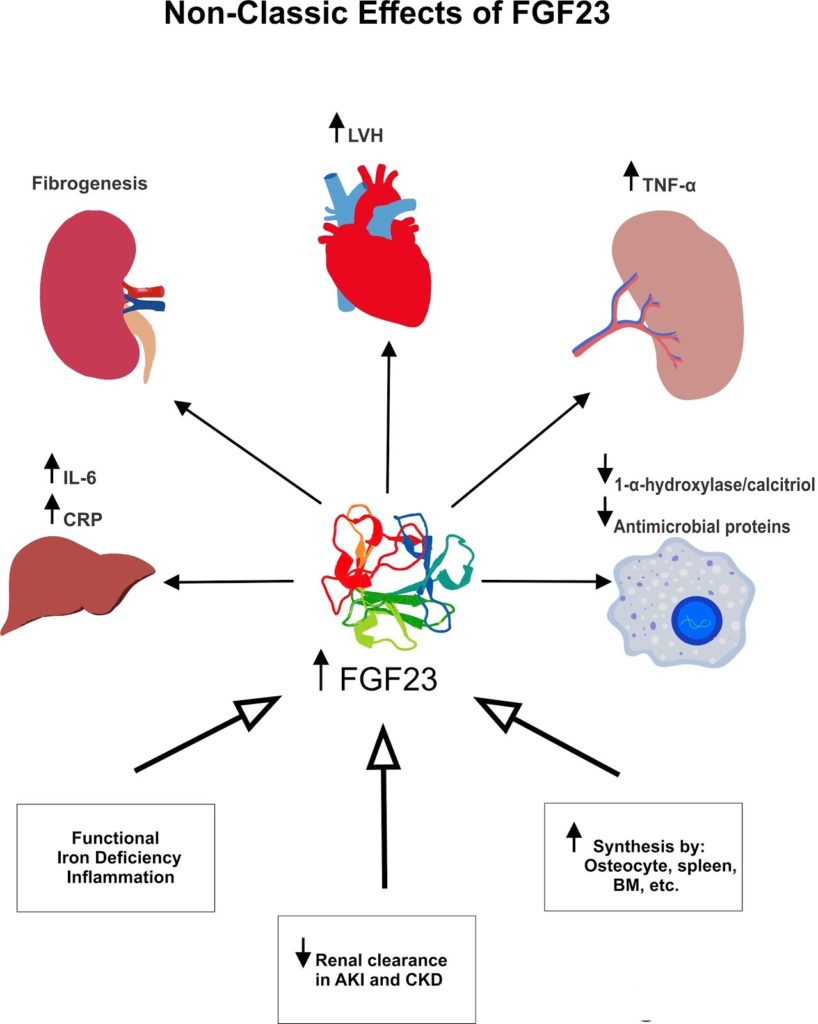
Non-classic (‘off-target’) effects of FGF23 in different tissues. Serum FGF23 increase in the setting of functional iron deficiency anemia, inflammation, reduced glomerular filtration rate and extra-osseous synthesis.
FGF23: Fibroblast Growth Factor 23, LVH: Left Ventricular Hypertrophy, TNF: Tumor Necrosis Factor, CRP: C- reactive peptide, BM: Bone Marrow, AKI: Acute Kidney Injury, CKD- Chronic Kidney Injury
FGF-23 and susceptibility to infections
In a murine model of E. coli pneumonia and CKD, Rossaint et al. demonstrated that an excess of FGF23 could impair the recruitment of polymorphonuclear cells (PMNs) by compromising integrin activation and migration to the site of infection. Similar effects have been found in AKI models and are thought to be related to immune dysregulation. However, these findings have been challenged by target engagement assays suggesting that FGFR2 may not be a target for FGF23.
Among other mechanisms, FGF23 can decrease the expression of 1-α hydroxylase in monocytes and decrease the activation of calcitriol; a hormone with known immunomodulatory effects. For example, calcitriol is involved in the maturation of monocytes to macrophages through nuclear vitamin D receptors. In addition, downregulation of the gene CYP27B1 (encodes 1-α hydroxylase) leads to reduced levels of cathelicidin; an antimicrobial protein involved in innate immunity. Translated in clinical studies, there is increasing evidence suggesting a link between FGF23 and susceptibility to infections. For example, high circulating FGF23 levels have been independently associated with the risk of infection-related hospitalization in a cohort of community-dwelling elderly individuals.
FGF23 as a prognostic marker of AKI and adverse outcomes
Serum FGF23 levels rise early in the onset of AKI, as seen in animal models of folic acid nephropathy. Furthermore, marked serum elevations of iFGF23 and cFGF23 in the later experiment resemble those reported in CKD models in mice. More recent studies have reported that serum FGF23 concentrations also increase in AKI due to other etiologies including infections, hemorrhage, urinary obstruction, and nephrectomy. Leaf DE et al. assessed serum cFGF23 in 250 patients undergoing cardiac surgery. They found that high cFGF23 levels at the end of cardiopulmonary bypass were independently associated with severe AKI, AKI-requiring renal replacement therapy and death. Similar findings are seen when urinary cFGF23 was measured in a more heterogeneous population of critically ill patients. This association was independent of other abnormal bone mineral disease parameters such as PTH, calcitriol, and phosphate, suggesting that FGF23 per se may be involved in more severe forms of AKI and poorer outcomes. However, further human epidemiological studies are needed. Previous reports in this field have been limited by different factors including small sample size, type of design (mostly observational), lack of ethnic diversity, etc.
FGF23 has also been investigated as a maker of disease progression in different cohorts of CKD patients. In a prospective study of patients without diabetes with an estimated glomerular filtration rate (eGFR) > 30 ml/min/1.73 m2, FGF23 predicted progression of CKD after adjustment for age, gender, GFR, proteinuria, and bone mineral parameters. The same correlation has been found in patients with CKD due to other etiologies including Ig A nephropathy, diabetes mellitus, etc.
The link between FGF23 increments and progressive CKD was tested in a large community-based study that included 13 448 participants from the Atherosclerosis Risk in Communities Study whose mean eGFR was 97 ml/min/1.73 m2. After a median follow-up of 19 years, 2% of the participants developed ESRD. When the highest FGF23 quintile (>54.6 pg/ml) was compared with the lowest quintile (<32.0 pg/ml), the authors found that higher FGF23 levels were associated with incident ESRD (hazard ratio, 2.10; 95% confidence interval, 1.31 – 3.36; P<0.001), after adjusting for confounders. Additional studies have demonstrated increased mortality due to cardiovascular complications, and all cause-of-death among patients with advanced CKD/ESKD.
In summary, FGF23 is a phosphaturic hormone that may play an important role in the pathophysiology and clinical course of AKI/CKD. Non-classic effects such as exacerbated inflammation/shock, higher infection risk, or toxic effects in the cardiovascular system could ultimately lead to adverse outcomes in patients with AKI/CKD, as suggested by epidemiological studies. Moreover, FGF23 offers new perspectives on the potential relationship between AKI and CKD. Further human studies exploring the FGF23-klotho axis in AKI/CKD are needed, as it offers a unique target for novel therapies that can improve renal and non-renal outcomes.
Acknowledgment: The author would like to express his gratitude to Dr. Pranav Garimella, Dr. Javier A. Neyra and Dr. Bellorin-Font for their valuable recommendations, and Isaac Philip for his artistic support with the figure above.
George Vasquez-Rios, MD
Internal Medicine Resident
Saint Louis University

