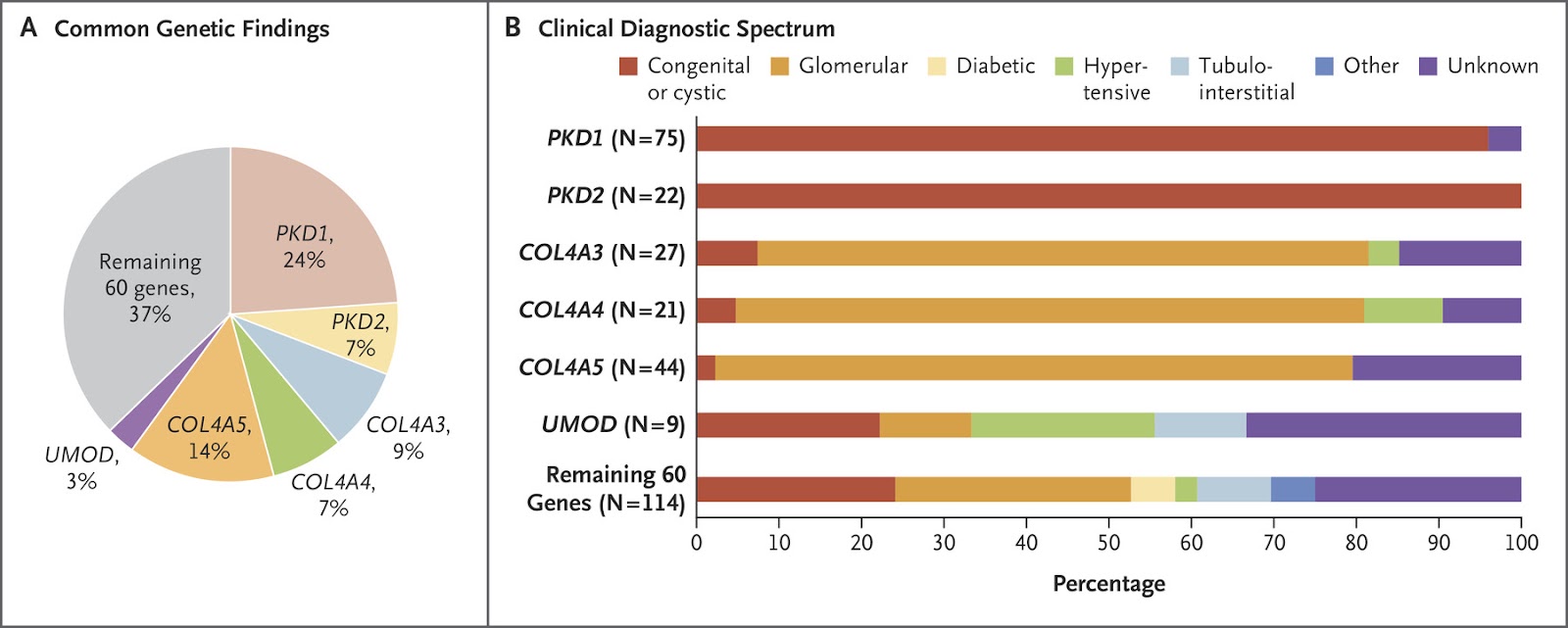
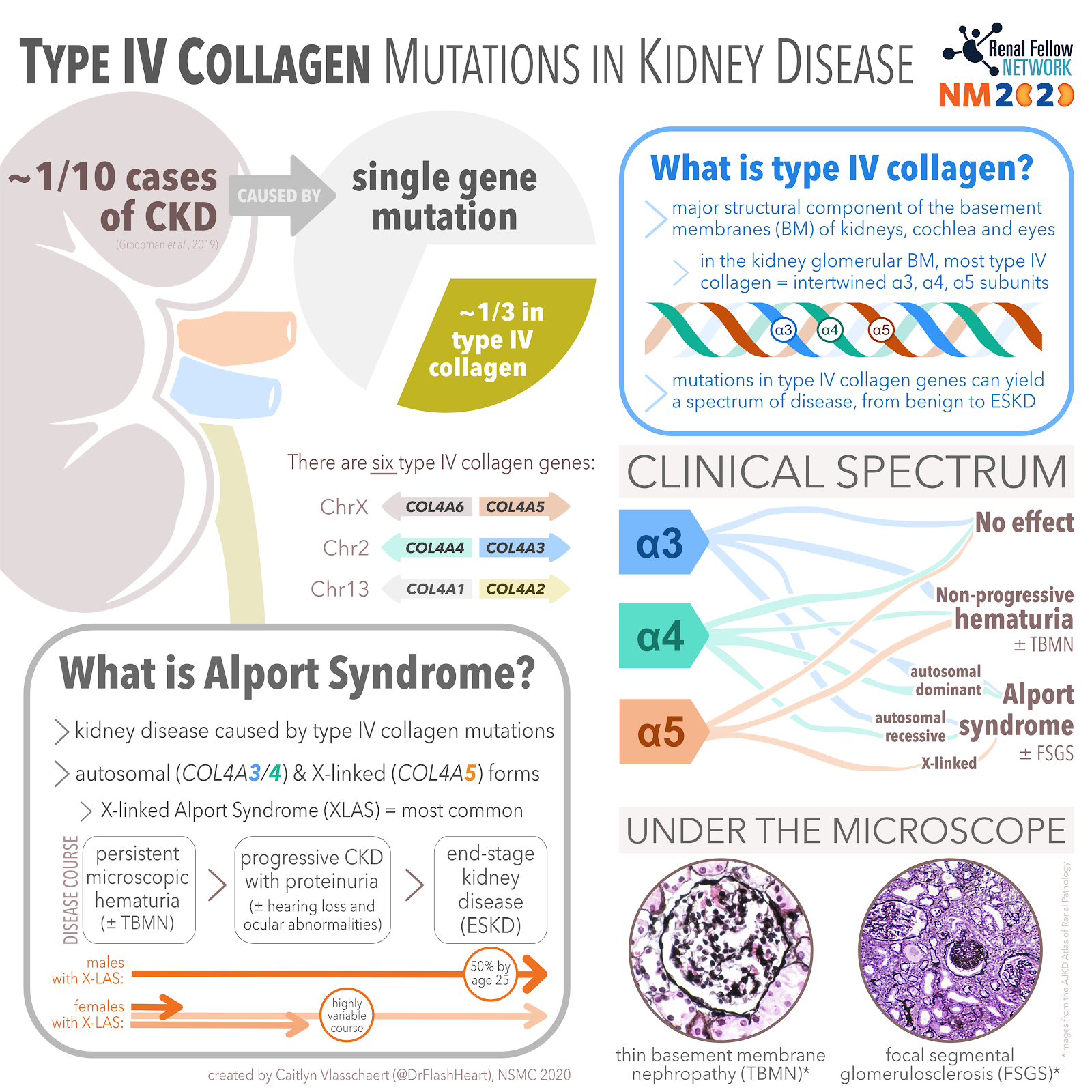
One in every 10 cases of chronic kidney disease (CKD) may be attributable to a single gene mutation. Read that again. One in every 10 cases. Wow. That was a shock to me. It took me a few times to fully comprehend that statement. A very interesting paper by Groopman et al. performed whole exome sequencing of a primarily adult (92%) cohort of over 3,000 patients with CKD (65% had already progressed to end stage kidney disease, or ESKD). Their study, discussed in NephJC, yielded a new genetic diagnosis in 9.3% of cases. Mutations in type IV collagen made up a third of this total (see Figure 1). Collagen mutations are classically associated with Alport syndrome. However, modern genetics has enabled us to recognize the compounding role of type IV collagen mutations in the pathogenesis of other causes of CKD including diabetic kidney disease and focal segmental glomerulosclerosis.
What is type IV collagen?
Collagen is the body’s main structural protein, the glue that holds all of our pieces together. There are over 30 types of collagen, but the predominant form (>90%) is Type I, which forms tendons and ligaments, and is found in all organs, blood vessels, and bones. Kidney fibrosis is primarily caused by deposition of type I collagen filaments.
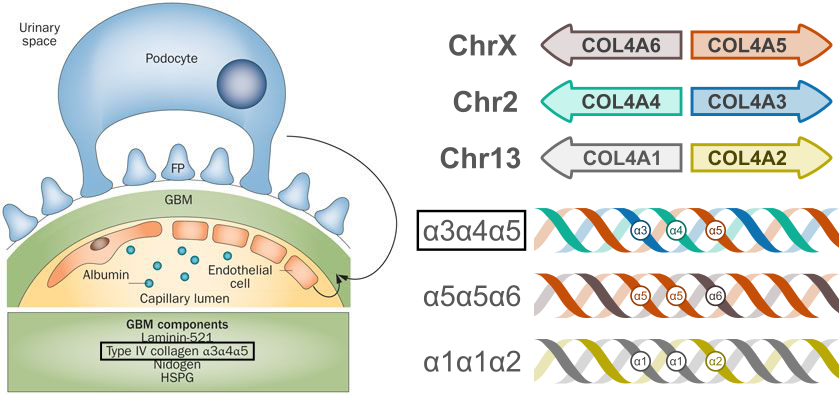
Type IV collagen is different— instead of forming tendon-like strands, it organizes into lattice-like networks that form the basement membranes of the kidney, cochlea and eyes. Most of the type IV collagen in the glomerular basement membrane is formed by intertwined α3, α4, and α5 subunits (Figure 2). Each of these subunits are transcribed from a different gene. The α5 subunit, derived from the COL4A5 gene on the X-chromosome, is mutated in X-linked (i.e., classic) Alport syndrome. Different mutations in the α3 or α4 subunit genes can lead to autosomal dominant or autosomal recessive forms of Alport syndrome, as well as thin basement membrane nephropathy (TBMN). However, the spectrum of kidney dysfunction caused by collagen gene mutations extends beyond these classically defined syndromes, as we will learn.
Below is a summary of the spectrum of kidney diseases known to be caused by Type IV collagen mutations:
1) Alport syndrome
Persistent microscopic hematuria is the earliest sign of Alport syndrome, typically present at birth. By the second decade of life, most males with X-linked Alport syndrome (XLAS; remember the mutated COL4A5 gene is located on the X chromosome) develop progressive CKD with proteinuria as well as sensorineural hearing loss and ocular abnormalities. Half of men reach end stage kidney disease (ESKD) by age 25, 90% by age 42 and 100% by age 60. Females with a heterozygous COL4A5 mutation have a milder disease course, typically without ESKD or hearing loss— although they may too develop advanced CKD and even ESKD. Check out the NephMadness 2020 Genetics Region to learn more about XLAS in women.
The primary treatment goal in Alport syndrome is to delay the requirement for kidney replacement therapies. Management of hypertension and proteinuria via blockade of the renin-angiotensin system is the current standard of care. Typically, angiotensin converting enzyme inhibitors (ACEis) or angiotensin receptor blockers (ARBs) are started when overt proteinuria is present (or even earlier). Targeted therapies are the focus of ongoing trials. All first degree relatives of patients with Alport syndrome should undergo a screening urinalysis and receive guidance on genetic testing.
2) Thin basement membrane nephropathy
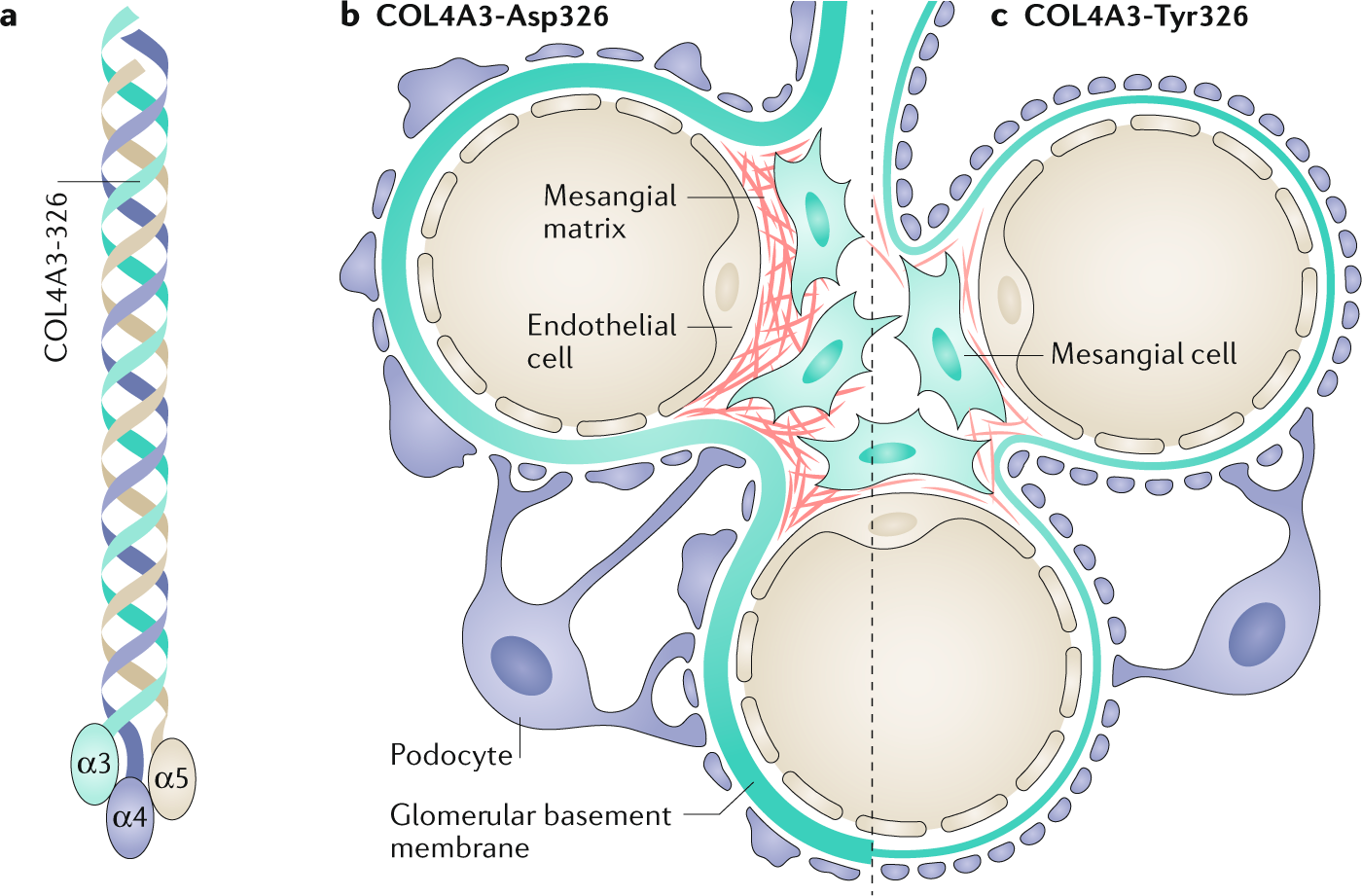
Within the spectrum of genetic hematuria syndromes is an entity called thin basement membrane nephropathy (TBMN). As the name implies, TBMN is a pathologic descriptor for a glomerular basement membrane that is about half its normal thickness. Historically, we used the term “thin basement membrane disease” to describe a benign hereditary condition characterized by thin basement membranes and persistent hematuria without any progressive kidney dysfunction or proteinuria, seen in patients with heterozygous (single allele) mutations in COL4A3 or COL4A4. However, we now know that the prognosis of TBMN is not always benign: a recent systematic review showed that 29% of people with heterozygous COL4A3/4 mutations develop CKD and 15% progress to ESRD. Similarly, females with single COL4A5 mutations and TBMN on microscopy might experience a clinically benign course or go on to develop kidney failure. Given the spectrum of phenotypes encountered with heterozygous COL4A3/4/5 mutations, exactly where TBMN fits under the umbrella of Alport syndrome is controversial (see the NephMadness 2020 Genetics Region for more). Understanding how genotype influences phenotype in this context is clinically important, for example when prognosticating for genetic counselling and assessing kidney donor candidacy. A recent position paper by the Alport Syndrome Classification Working Group suggested that all patients with heterozygous COL4A3 or COL4A4 mutations should cautiously be diagnosed with Alport syndrome.
3) Type IV collagen mutations in FSGS
Secondary FSGS commonly develops late in the course of Alport syndrome. More recently, we’ve learned that some type IV collagen mutations also appear to lead to a primary FSGS phenotype, sometimes even in the absence of hematuria. Two thirds of the patients found to have COL4A3, COL4A4, or COL4A5 pathogenic variants in the Groopman study (Figure 1) actually had pre-existing light microscopy-based diagnoses of FSGS but not Alport syndrome or TBMN. These variants may explain up to 38% of familial FSGS and 3% of sporadic cases, which has significant implications for the workup and treatment of primary FSGS (see this great Tweetorial by Justin Davis for more on this).
4) Type IV collagen in diabetic kidney disease
Type IV collagen seems to influence how diabetes affects the kidneys. Different variants in the α3 subunit gene can either protect the kidneys from or enhance the progression of diabetic kidney disease (DKD) in individuals with type 1 diabetes mellitus and with maturity-onset diabetes of youth (MODY). GWAS studies of DKD in type 2 diabetes have not elicited associations with collagen gene mutations to date, but type IV collagen turnover appears important in its pathogenesis. Type IV collagen is a major constituent of the expanded GBM and mesangium seen in DKD. In several small studies, DKD progression has been correlated with urinary type IV collagen levels, and its use as a biomarker may better predict kidney failure than quantitative albuminuria.
5) Type IV collagen mutations in cystic kidney disease
Autosomal dominant polycystic kidney disease (ADPKD; caused by mutations in PKD1 or PKD2) is the most common heritable cause of kidney cysts. Type IV collagen mutations have been found in patients with bilateral kidney cysts and normal ADPKD genes. Interestingly, like COL4A3/4/5 mutations, PKD1/2 mutations (without clear ADPKD phenotype) accounted for one third of new genetic diagnoses in the Groopman et al. study (Figure 1). Clearly, our understanding of the clinical spectrum of disease caused by mutations in these classic kidney disease genes is rapidly evolving.
6) Kidney disease due to mutations in the other Type IV collagen subunits
Most type IV collagen found in the glomerulus is comprised of α3, α4, and α5 subunits forming a triple helix. We have learned about how mutations in the genes coding for these subunits cause glomerular kidney disease. There are actually six type IV collagen subunits in total; COL4A1 through COL4A6 (Figure 2). In addition to the α3α4α5 helix, α1α1α2 and α5α5α6 helices also form parts of basement membrane networks. Mutations in the α1 and α2 subunits have some reported associations with kidney pathology, including isolated and syndromic forms of kidney disease. There are no specific kidney diseases associated with α6 subunit mutations, except for a specific form of XLAS caused by contiguous deletion of COL4A5 and COL4A6 (these genes are adjacent to one another on chromosome X; see Figure 2).
Caitlyn, Vlasschaert, MD MSc.
Internal Medicine Resident
Queen’s Internal Medicine, Kingston, Ontario, Candada
NSMC Intern 2020
@DrFlashHeart
Acknowledgements: Thank you to Drs. Matthew Sparks (@nephro_sparks), Paul Phelan (@paulphel), Samira Farouk (@ssfarouk), Michelle Rheault (@rheault_m) and Matt Lanktree (@MattLanktree) for their input.


Lucid, Updated and incredibly creative visuals. Great going.