The close relationship between extracellular (free) calcium concentration and parathyroid hormone (PTH) is tightly sensed by the calcium-sensing receptor (CaSR) expressed at the membrane of parathyroid cells. This control is so well regulated that even a small increase that approaches the upper limit of normal could potentially completely suppress PTH secretion. Therefore, when evaluating a patient presenting with hypercalcemia, measuring a concomitant PTH level is a key first step: if suppressed, extra-parathyroid etiologies should be considered (including hypercalcemia of malignancy mediated by PTHrP or osteolytic bone metastases, vitamin D intoxication, and chronic granulomatous disease); if not suppressed, primary hyperparathyroidism (PHPT) and familial hypocalciuric hypercalcemia (FHH) should be considered. Even if the precise epidemiology of FHH is still unknown, it is much rarer than PHPT which is very common and often underdiagnosed.
PHPT is typically diagnosed on the basis of hypercalcemia in the presence of an elevated PTH concentration. Single adenomas account for 80-85% of cases, double adenomas an additional 2 to 5%, and glandular hyperplasia approximately 6%. Very rarely (<1% of cases), a carcinoma could be the cause. When more than one adenoma is found, a genetic disorder (such as multiple endocrine neoplasia or a jaw tumor syndrome) should be suspected. Regardless of the underlying etiology, there is abnormal regulation of PTH secretion by calcium. Sustained PTH elevation stimulates bone remodeling, particularly osteoclastic resorption, releasing calcium and phosphate which are then excreted into urine. The elevation in PTH also results in increased intestinal reabsorption of calcium mediated by increased production of calcitriol, and in increased renal tubular reabsorption of calcium. Thus, the main complications of PHPT are in the kidney (nephrolithiasis ± chronic kidney disease) and/or bone (osteopenia ± fragility fractures in the vertebrae, hip, and wrist). Currently (at least in developed countries), symptomatic hypercalcemia due to PHPT represents a rare finding but subclinical nephrolithiasis and vertebral fractures may be found in at least 50% of patients. Parathyroidectomy is indicated for all symptomatic and asymptomatic patients with evidence of end-organ damage or progression.
FHH was first described by Foley et al. in 1972 as an autosomal dominant genetic disorder due to a defect of extracellular calcium sensing in the parathyroid glands as well as in the kidneys. Three loss-of-function mutations have been reported to cause FHH: CASR, encoding the CaSR itself in FHH1; GNA11, encoding the Gα11 protein in FHH2; and AP2S1, encoding the adaptor-related protein complex 2, sigma-2 subunit in FHH3. As a result of these mutations, the set-point at which extracellular calcium concentration is sensed and stimulates/inhibits PTH secretion is shifted to a higher level (Figure 1). Even if small cohorts of such patients exist around the world, very few of them are symptomatic and fewer still present with complications. In most cases, parathyroid gland surgery does not cure FHH and must be avoided.
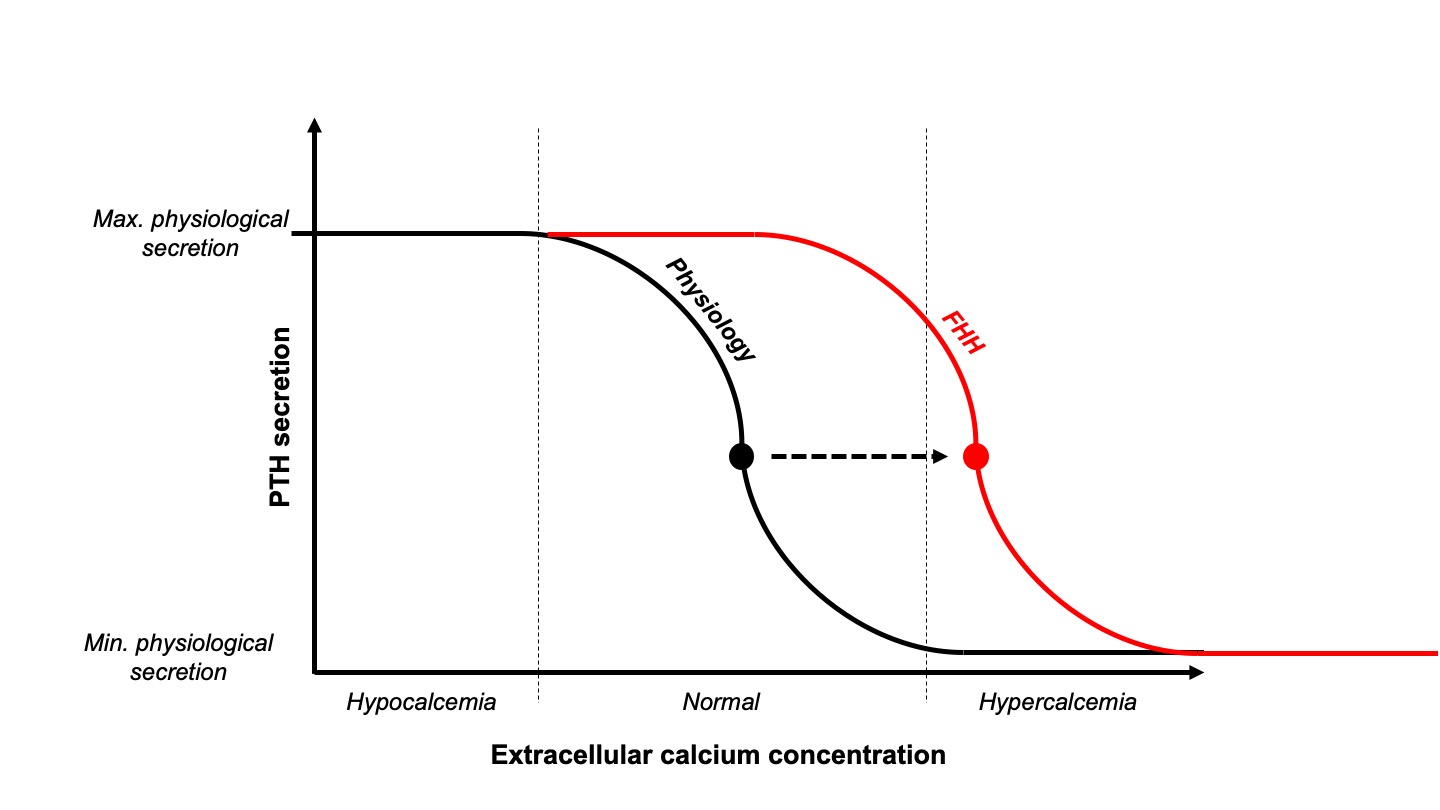
Distinguishing between these two diseases is essential as FHH usually has a benign natural history with patients rarely developing bone or kidney complications, and parathyroidectomy is generally unhelpful. PHPT is usually diagnosed in older women (over men); in comparison, FHH patients are usually younger and may be male or female. Even if the PTH level and hypercalciuria are usually lower in FHH than in PHPT patients, there is a large overlap, making use of these parameters alone unreliable for distinguishing diagnoses. Plasma PTH concentration is within the normal range in 10-50% patients with PHPT and in about 80% of patients with FHH. The latest guidelines on the diagnosis of PHPT state that calcium-to-creatinine clearance ratio calculated from 24-h urine collection (UCCR) can help distinguish between FHH and PHPT: UCCR is typically greater than 0.02 in PHPT patients and lower than 0.01 in FHH patients. Unfortunately, UCCR values can also overlap in FHH and PHPT patients and genetic testing may be required to distinguish these conditions.
To help tackle this diagnostic dilemma in patients with hypercalcemia and normal PTH levels, a new easy-to-use bedside tool called Pro-FHH has recently been developed. This tool additionally accounts for the circulating concentration of bone remodeling markers such as alkaline phosphatase and/or osteocalcin, Pro-FHH performed better in differentiating FHH patients from PHPT ones, requiring less genetic tests by adding only one bone remodeling marker measurement into a probability score equation. Using Pro-FHH (the tool can be downloaded here) could prevent unnecessary surgery in FHH patients. However, the diagnostic accuracy of the tool remains to be tested in patients with elevated PTH values.
In conclusion, FHH is a rare disease that can mimic PHPT and although differentiation can be challenging, consideration of this alternative diagnosis and use of tests such as UCCR and Pro-FHH may help prevent unnecessary surgery.
Post by: Jean-Philippe Bertocchio

and now you can use the dedicated on-line tool : http://www.profhh.org in EN 🇺🇸 and FR🇫🇷
It is great and simplified article .. I will share and re publish it in my scientific group ( port said neonatology group)..