Amyloidoses are a group of diseases resulting from deposition of amyloid, insoluble fibrils derived from various precursor proteins, into extracellular tissues. Amyloidoses are acquired or hereditary, and depending on where amyloid deposits, can affect a wide range of organs systems, including the kidneys, heart, liver, gastrointestinal tract, peripheral nerves. The initial step in formation of an abnormal amyloid protein is the misfolding of a precursor protein. Over 30 proteins have been identified as amyloidogenic, but mass spectrometry has identified many more proteins with the potential to form amyloid in humans. The misfolded proteins then aggregate and form fibrillar material that arranges into the characteristic β-pleated sheet seen in amyloidosis.
The classification of amyloidosis is based on the precursor protein from which the fibrils are formed. The major types of systemic amyloidosis are: Ig light chain (AL), amyloid A (AA), hereditary amyloidosis [ATTR (transthyretin), AFib (Fibrinogen A α-chain), ALys (lysozyme), AApoAI (Apolipoprotein AI), AApoAII (Apolipoprotein AII), AGel (gelsolin), ACys (cystatin C)], age-related (senile) systemic amyloidosis, ALect2, and β2-microglobulin (β2m) amyloidosis. The protein fibrils of all these types of amyloidosis are 8-12 nm in diameter, similar in structure, and contain several non-fibrillary components including glycosaminoglycans (GAGs) and serum amyloid P component (SAP), which stabilize and protect the fibrils from proteolysis. The β-pleated sheet formation of fibrils gives them the ability to bind Congo red dye (seen on light microscopy) in an organized, intercalated way that emits apple-green birefringence under polarized light. This birefringence is the most widely available gold standard for diagnosis of amyloidosis, however neither light nor electron microscopy can distinguish different types of amyloidosis. Diagnosis of amyloid type occurs via immunohistochemistry, mass spectrometry, or amino acid sequencing of proteins in amyloid deposits.
Amyloid can deposit in almost any organ system, leading to a wide range of clinical presentations. While the clinical manifestations of amyloidosis are rarely specific to type of amyloidosis, the kidneys are most commonly involved in AL, AA, AFib, ALect2, and AApoA1 amyloidoses. Amyloid can deposit in the glomerulus (predominant), tubulointerstitium, or vasculature of the kidney. A summary of the pathologic findings common to amyloidoses is shown in Table 1. Kidney disease usually presents as reduced glomerular filtration rate (GFR) and/or proteinuria which may progress to nephrotic syndrome. Without treatment, GFR may progressively decline and patients may develop end-stage kidney disease (ESKD). Here, we review some of the clinicopathologic and distinguishing features of some of the most common amyloidoses.
Table 1. Summary of common pathologic findings in amyloidosis with kidney involvement.
| Deposits | Pathology |
| Light microscopy | 1. Amorphous pink material in mesangium, peripheral capillary walls, tubulointerstitium, and arteriolar walls. 2. Glomeruli may show a nodular sclerosis pattern mimicking diabetic nephropathy. 3. Arteriolar deposits may mimic hyalinosis. |
| Special stains | 1. Amyloid deposits are weakly periodic acid-Schiff (PAS) positive and silver negative (unlike Kimmelstiel-Wilson nodules in diabetes and hyaline deposits in arterioles which are both positive for PAS). 2. Trichrome shows a dull grey appearance in the deposits (in contrast to the normal bright blue staining of collagen). 3. Congo red stain shows orange-colored positivity by regular microscopy and apple-green birefringence under polarized light. |
| Electron microscopy | 1. Randomly arranged fibrils measuring 8-12 nm in diameter. 2. Amyloid fibrils characteristically have an “invasive” appearance, growing through glomerular capillary walls causing spike-like remodelling of the capillary walls (so-called “cocks-comb” appearance). This can sometimes be seen by silver stain on light microscopy. |
AL Amyloidosis
Amyloid protein– AL (monoclonal Ig light chains)
Pathogenesis– Clonal plasma cells make monoclonal light chains. Plasma cell dyscrasias leading to AL amyloidosis may range from monoclonal gammopathy of undetermined significance (MGUS) to multiple myeloma (MM).
Distinguishing characteristics– AL amyloidosis is the most prevalent type of amyloid disease in most parts of the United States (US). AL fibrils are most often lambda light chains (four times more likely than kappa light chains). This is in contrast to MM, in which kappa light chains are more predominant. Frequent presenting symptoms are peripheral neuropathy and carpal tunnel syndrome. Involvement of other soft tissues (macroglossia, muscular pseudohypertrophy, salivary gland enlargement) is unique to AL amyloidosis.
Treatment– Chemotherapy (e.g. melphalan) targeting clonal plasma cells and autologous stem cell transplant (ASCT) are mainstays of therapy for AL amyloidosis. Bortezomib is a proteasome inhibitor that has been shown to be effective in inducing a rapid treatment response in AL amyloidosis. Bortezomib-based regimens are recommended for patients who are not eligible for ASCT. Additionally lenalidomide and pomalidomide are immunomodulators that have more recently been implemented as therapy for refractory disease.
Case 1: Early renal amyloidosis, AL type (lambda), in a patient with multiple myeloma.
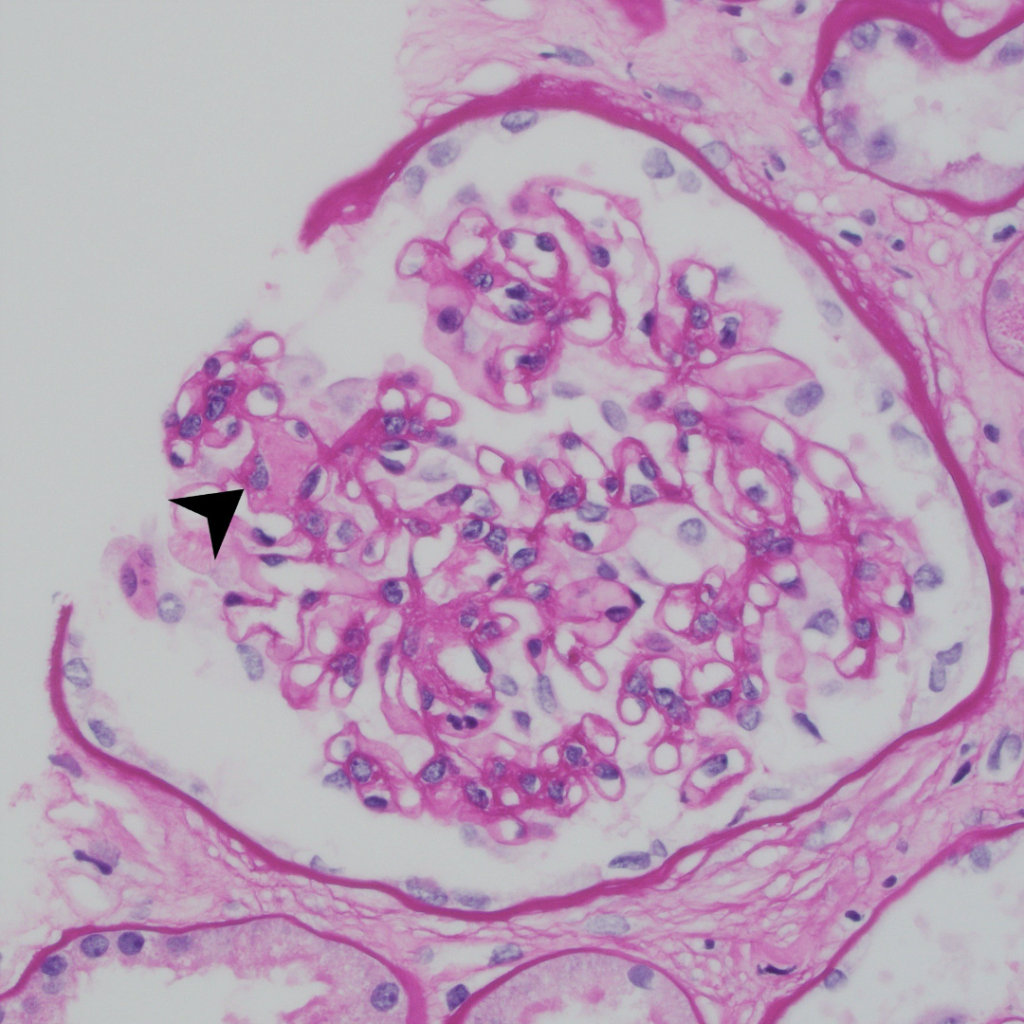
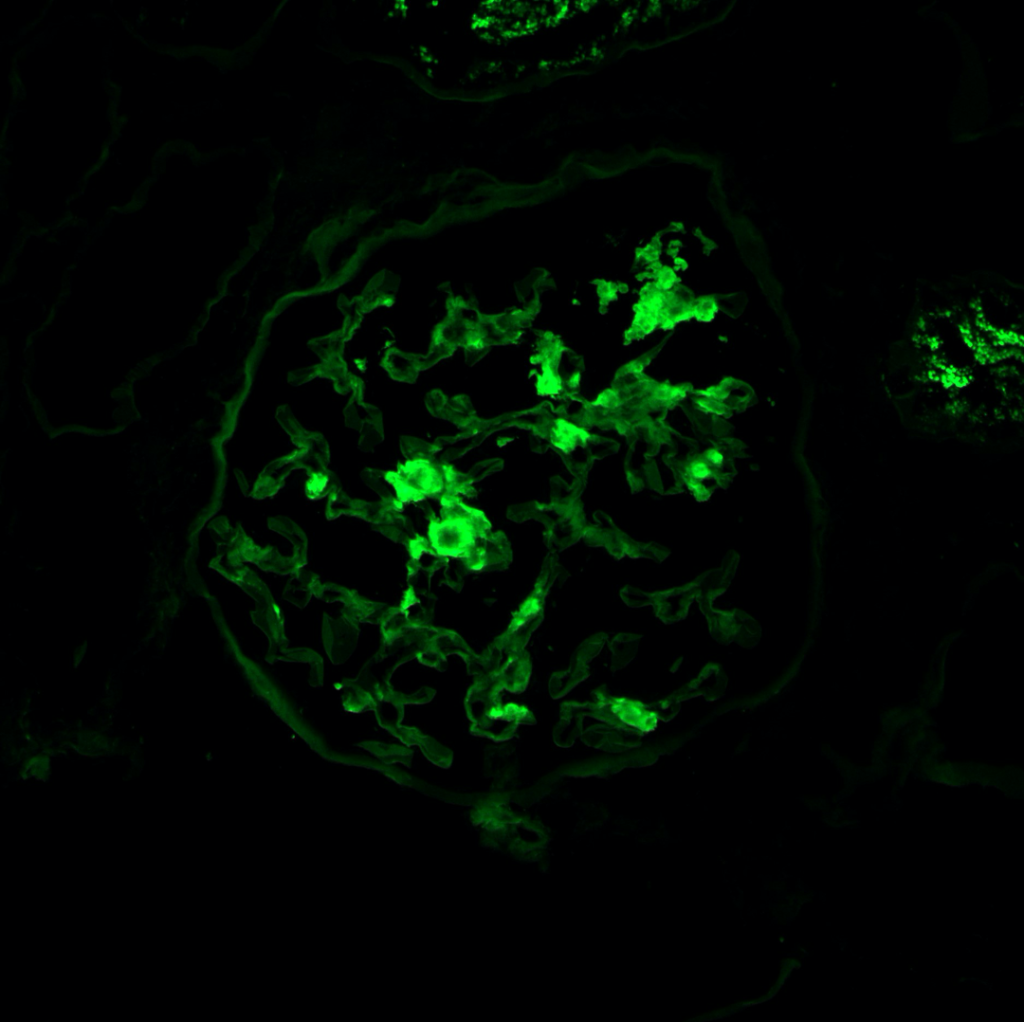
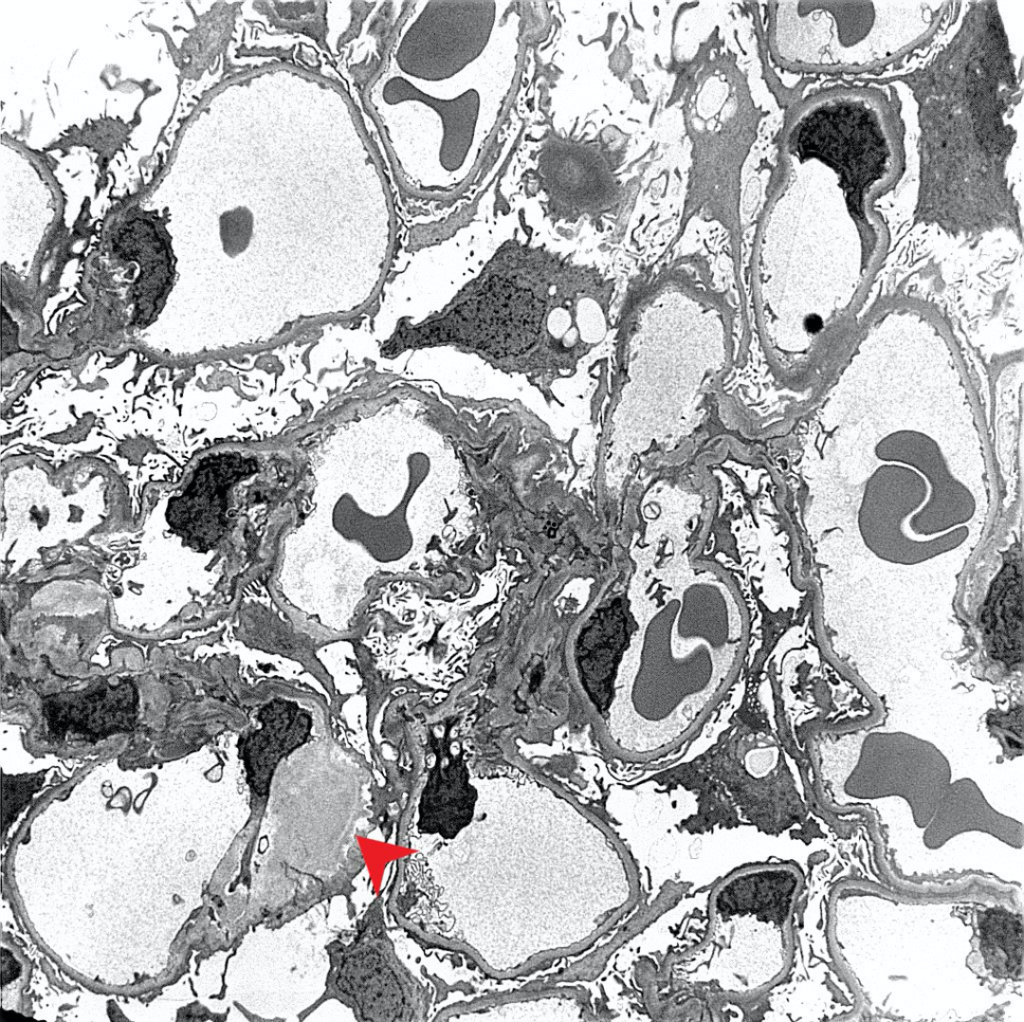
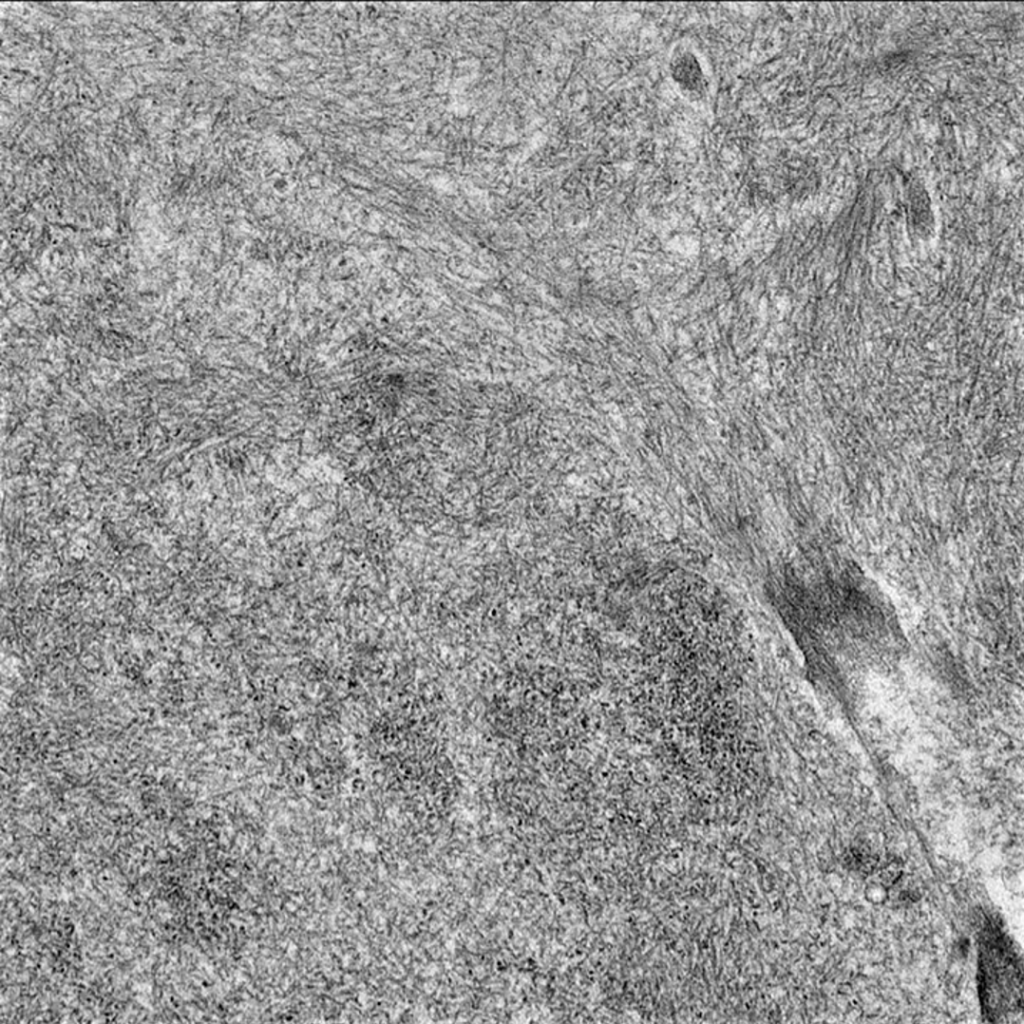
AA Amyloidosis
Amyloid protein– SAA (serum amyloid A)
Pathogenesis– Long-standing inflammatory disorders or infections (rheumatoid arthritis, familial Mediterranean fever (FMF), inflammatory bowel disease, and chronic infections) lead to overproduction of SAA, an apolipoprotein that is an acute-phase reactant. Sustained high amounts of SAA, posttranslational modifications, and intrinsic properties of the SAA protein may lead to protein misfolding and amyloidosis.
Distinguishing characteristics– AA amyloidosis is the second most common cause of systemic amyloidosis, behind AL amyloidosis. In some areas of the US, like San Francisco, AA amyloidosis may be the most common form. Proteinuria and kidney function decline are the most frequent presenting clinical manifestations.
Treatment– Given the diversity of etiologies, treatment of AA amyloidosis is primarily aimed towards reduction of SAA production by treating the underlying inflammatory disorder with antibiotics for chronic infections, colchicine for FMF, and immunosuppressive or biologic therapies (e.g. tumor necrosis factor inhibitors) for autoimmune diseases.
Case 2: An example of renal amyloidosis, AA type, in a patient with chronic hepatitis B.

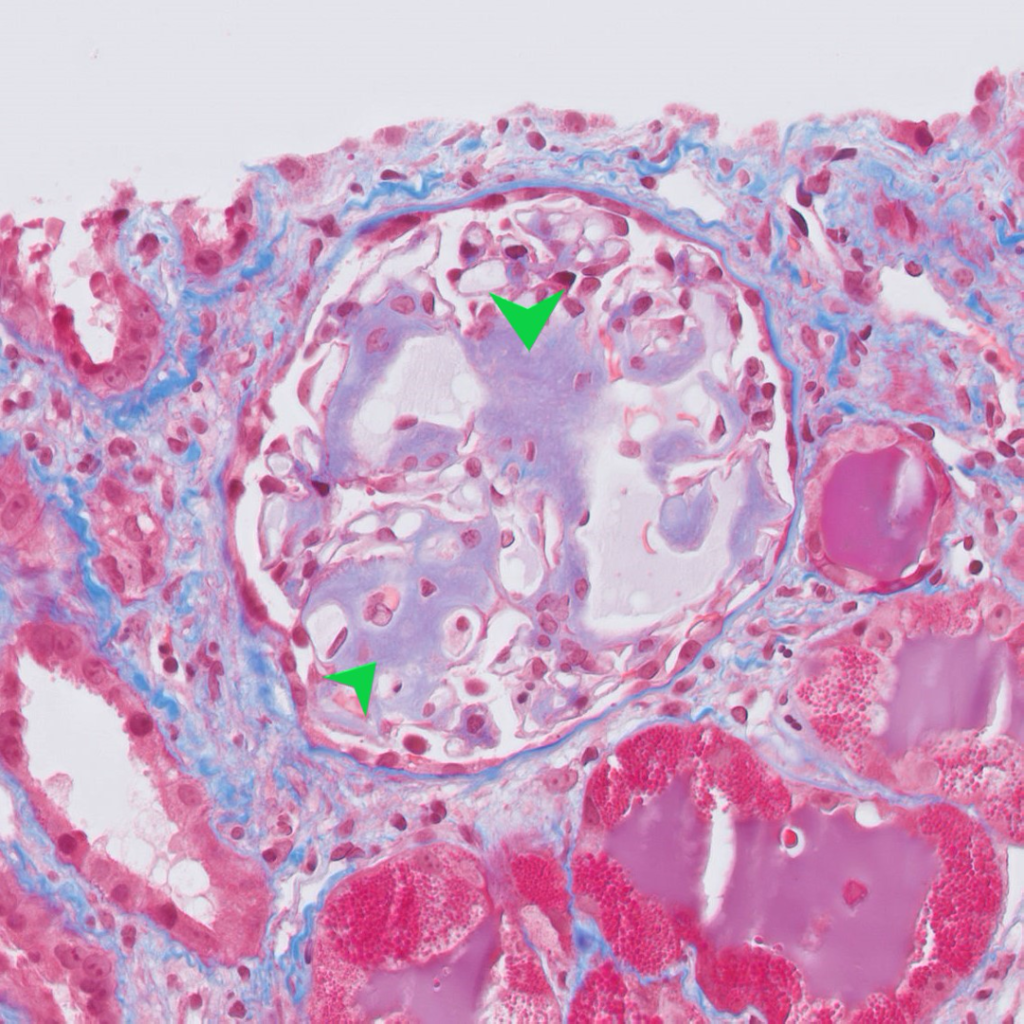
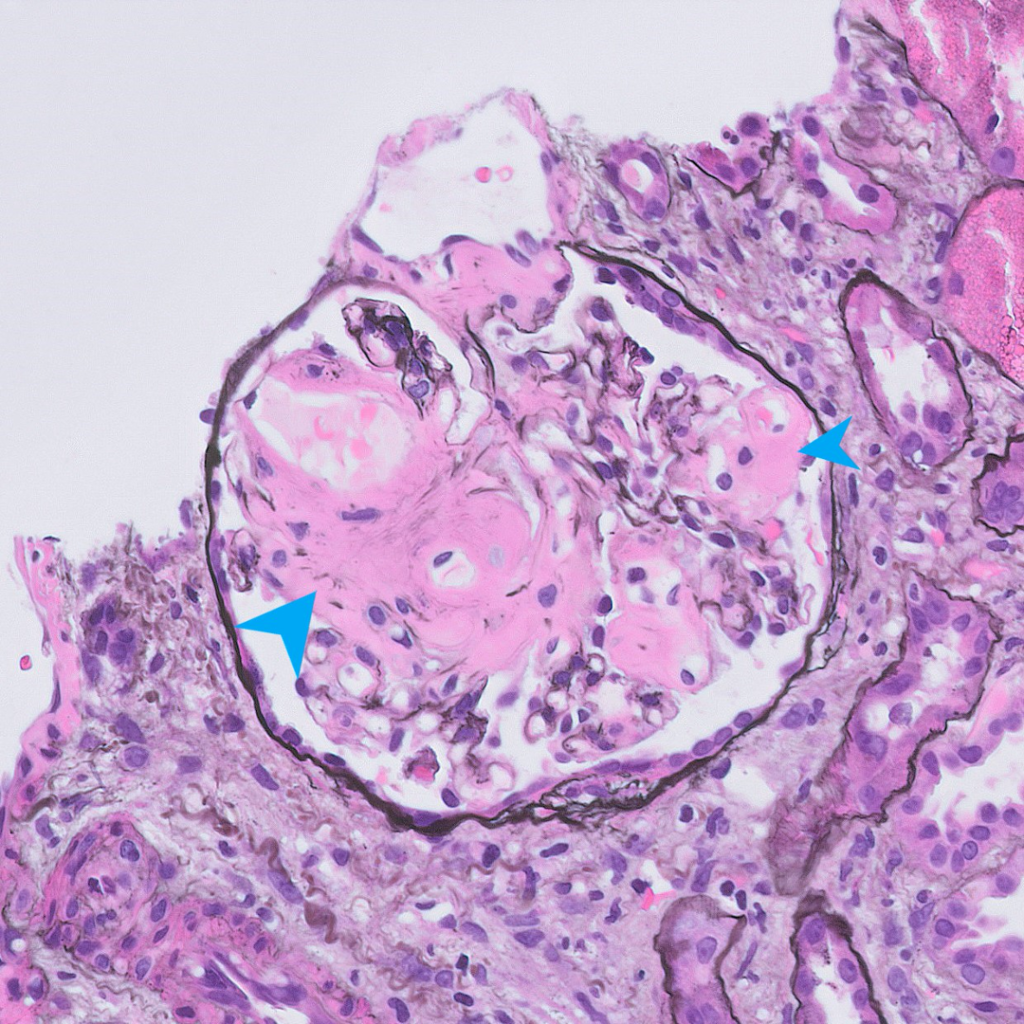
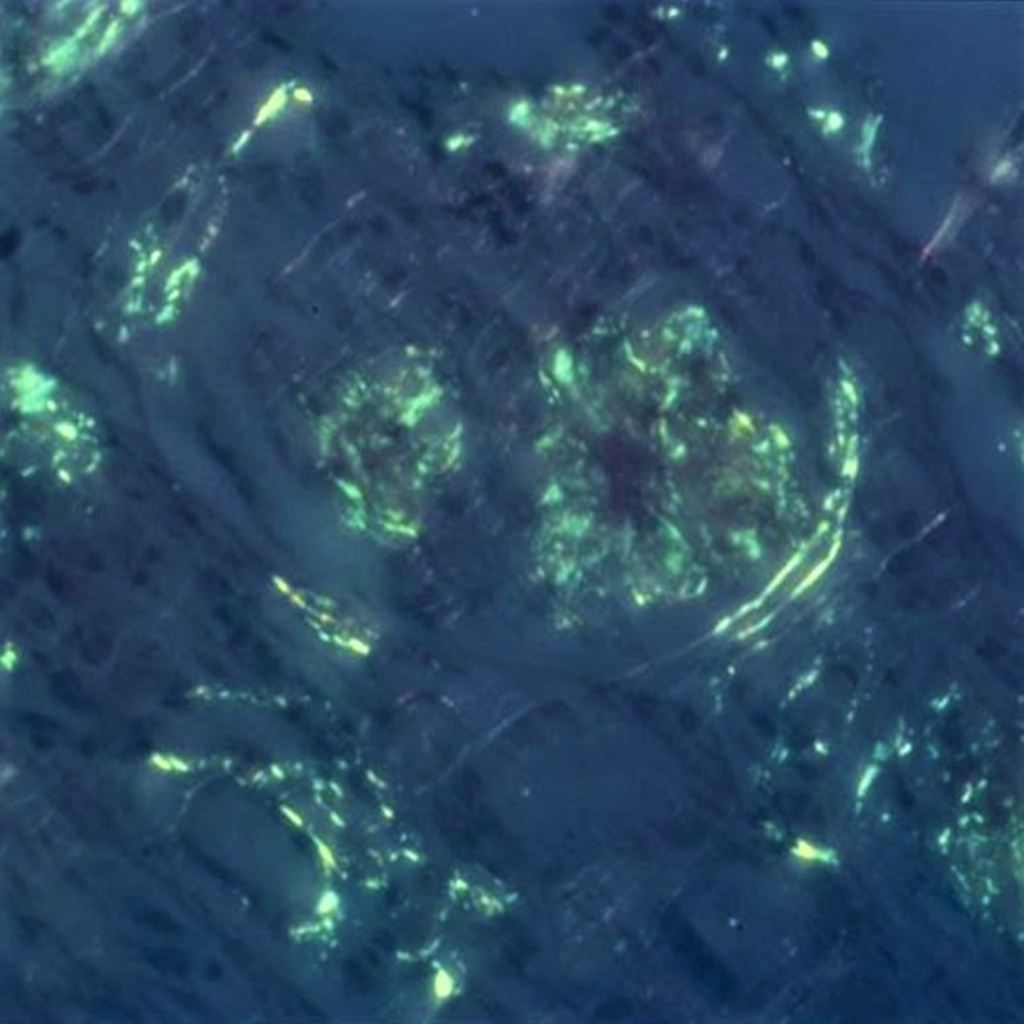
Figure 2d: Congo red stain, 400x. Congo red stain viewed under polarized light showing “apple green” birefringence. Picture courtesy Dr. Zoltan Laszik of UCSF Pathology.
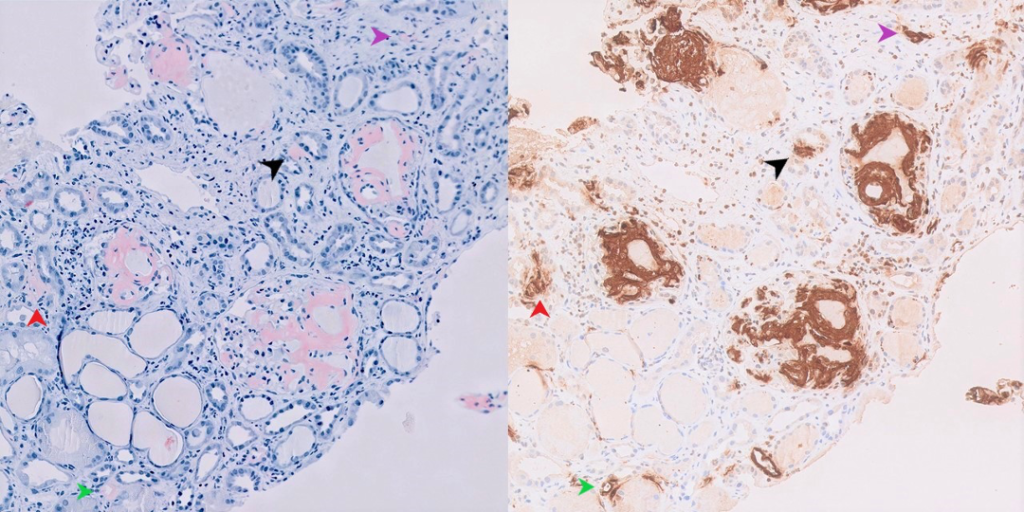
Hereditary Amyloidosis
Amyloid protein– ATTR (transthyretin), AFib (Fibrinogen A α-chain), ALys (lysozyme), AApoAI (Apolipoprotein AI), AApoAII (Apolipoprotein AII), AGel (gelsolin), and ACys (cystatin C)
Pathogenesis– Gene mutations in precursor proteins lead to misfolding and deposition as amyloid.
Distinguishing characteristics– AFib, AApoA1, ALys, and occasionally AGel amyloidoses can affect the kidney. Early and aggressive organ involvement may raise the possibility of a hereditary form of amyloidosis. These rare forms are often detected by mass spectometry only, as there are no reliable tissue stains for these types.
Treatment– Orthotropic liver transplantation is the treatment of choice in select patients with hereditary ATTR, AApoAI, and AFib amyloidoses (these amyloidogenic proteins are mainly made in the liver). Treatment of other hereditary amyloidoses is mainly transplantation to replace the failing organ affected by amyloid deposits. Novel RNA-inhibiting agents that interfere with hepatic synthesis of TTR are undergoing active investigation as therapies for ATTR amyloidosis.
Age-related (Senile) Systemic Amyloidosis
Amyloid protein– ATTR (wild-type transthyretin)
Pathogenesis– Wild-type ATTR becomes structurally unstable with age, resulting in protein misfolding and formation of amyloid.
Distinguishing characteristics– Carpal tunnel syndrome is a common early symptom (also in hereditary ATTR amyloidosis). ATTR fibrils have a predilection for depositing in the heart, thus heart failure is a common presentation.
Treatment– Tafamidis is a compound that binds and stabilizes the transthyretin tetramer in the plasma and reduces formation of TTR amyloid. Unlike in hereditary ATTR amyloidosis, wild-type ATTR is not made in the liver, thus liver transplantation is not indicated.
ALECT2 Amyloidosis
Amyloid protein– Lect2 (leukocyte chemotactic factor 2)
Pathogenesis– Unknown
Distinguishing characteristics– ALECT2 amyloid is a slowly progressive disease that selectively affects the cortical interstitium of the kidney and the liver. Most patients present with minimal proteinuria and impaired renal function. Cardiac involvement rarely occurs, leading to better survival than in other amyloid types. Despite being a newly described amyloid type, ALECT2 amyloidosis is the third most common cause of acquired renal amyloidosis, occurring predominantly in patients from South Asia, North Africa, the Middle East, and Mexico.
Treatment– There is no specific treatment, but in the setting of advanced kidney disease, kidney transplant is an option. However, with ongoing synthesis of Lect2 by the liver, there is high risk for recurrence of disease. Recurrent and de novo forms of ALECT2 have been described in the setting of kidney transplantation.
β2-microglobulin (β2m) Amyloidosis
Amyloid protein– Aβ2m (β2-microglobulin)
Pathogenesis– Aβ2m is a component of major histocompatibility complex that is normally filtered through the glomerulus and reabsorbed and catabolized in the proximal tubules. When kidney function declines, clearance and catabolism of Aβ2m also declines, leading to accumulation and deposition in tissues. Exposure to synthetic materials during hemodialysis has also been hypothesized to stimulate Aβ2m production through complement-mediated mechanisms.
Distinguishing characteristics– Aβ2m has an affinity to joint tissues and often presents with carpal tunnel syndrome. Bone cysts containing amyloid can also be found on x-ray.
Treatment– With high flux dialysis membranes that provide good clearance of Aβ2m, this is becoming an increasingly more rare cause of amyloidosis.
In recent years, typing of amyloid fibrils have improved with the increasing use of mass spectrometry, cardiac magnetic resonance imaging, and scintigraphy. Survival in patients with AL amyloidosis, the most common type of amyloidosis, is improving with the implementation of novel chemotherapeutic regimens. Early diagnosis remains the key to improved outcomes. As therapies advance to include RNA inhibitors, fibril formation stabilizers and inhibitors, and targeted immunotherapy, clinical outcomes for patients with various types of amyloidoses will continue to improve, even for those with advanced organ involvement.
Debbie Chen
Nephrology Fellow
University of California-San Franscisco


Amazingly written and meticulously crafted. Thank you Debbie