Welcome to the 11th case of the Skeleton Key Group, a team of twenty -odd nephrology fellows who work together to build a monthly education package for Renal Fellow Network. The cases are actual cases (without patient identifying information) that intrigued the treating fellow.
Written by: Tiffany Truong
Visual Abstract by: Varun Mamidi
Special thanks to Dr. Mohammad Alsawah
A. The Stem
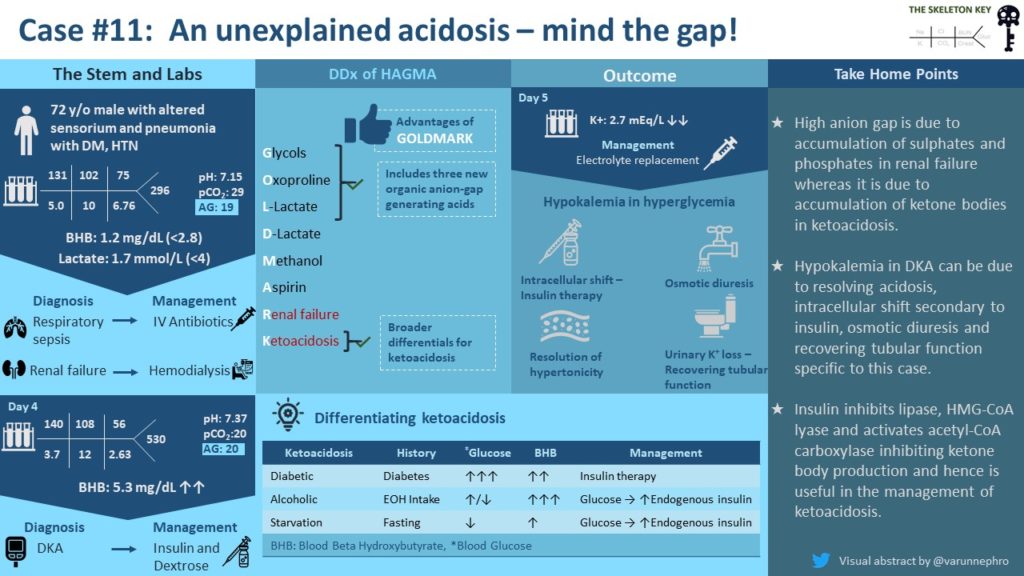
A 72 year-old male nursing home resident with a history of hypertension, diabetes mellitus, gout, and vascular dementia is admitted to the hospital with acute onset of agitation and disorientation. Home medications include aspirin, atorvastatin, metoprolol, amlodipine and colchicine. He was not on any medications for diabetes due to prior hypoglycemic events. He had no history of recent trauma or infection, and no fevers, incontinence, or cough.
On admission, his blood pressure was 93/62, which improved to 126/72 after infusion of 2 liters of normal saline.
Vitals– Temp 36.5°C| Blood Pressure 126/72 mmHg | Pulse 81 bpm | Resp 16/minute
Neurologic: No focal neurologic deficits. Agitated, on 4 point restraints
Head/Ears/Eyes/Nose/Throat: Atraumatic. No scleral icterus.
Cardiovascular: RRR, normal S1/S2, no murmurs.
Pulmonary: Right basilar crackles, no increased work of breathing
Abdominal: Soft, nondistended, nontender, positive bowel sounds
Musculoskeletal: No edema, no deformities
Dermatologic: Poor skin turgor. Dry mucous membranes. No rashes.
Imaging revealed right lower lung lobe opacities, and he was started on ceftriaxone and doxycycline.
B. The Labs
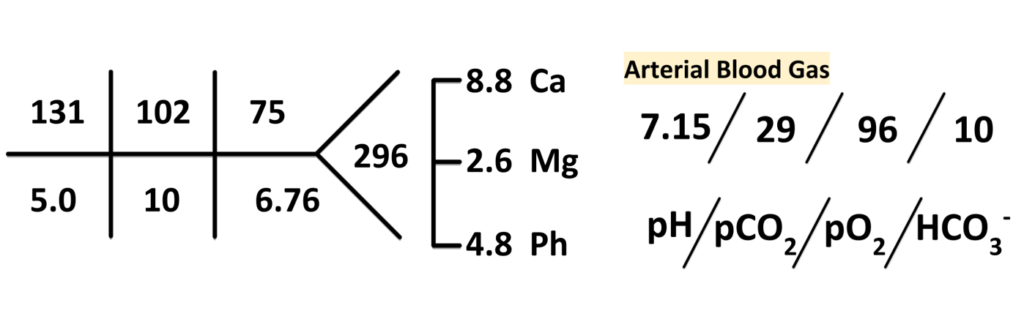
Lactic acid: 1.7 mmol/L.
Beta-hydroxybutyrate: 1.2 mg/dL (normal less than 2.8 mg/dL).
Urinalysis: no glucose, trace ketones.
Next day labs resulted with undetectable ethanol, methanol and ethylene glycol.
C. Differential Diagnoses
What is the cause of his acidosis? First, let’s evaluate his anion gap.
Anion gap = Na+ – (Cl– + HCO3–) so for this patient 131 – (102 + 10) = 19
Let’s review the differential diagnoses of a high anion gap metabolic acidosis (HAGMA). The differential for a HAGMA is commonly known by mnemonics, the historically used MUDPILES and more recently GOLDMARK introduced in 2008 in this article.
The Mnemonics of High Anion Gap Metabolic Acidosis (HAGMA)


Why should we switch from MUDPILES to GOLDMARK?
According to the authors who created the GOLDMARK mnemonic:
- Metabolic acidosis due to excessive paraldehyde or phenformin use has become very rare.
- Iron and isoniazid are just two of many drugs and toxins that can cause hypotension and lactic acidosis. Isoniazid can also generate a component of ketoacidosis.
- Three new organic acids and acid precursors have been recognized as causes of AGMA:
- 1) D-lactate, 2) 5-oxoproline, 3) propylene glycol
D. More Data
Our patient’s history and labs ruled out most causes of HAGMA, leaving kidney failure as his cause of metabolic acidosis. He received 2 ampules (1 ampule = 50 mEq NaHCO3 in 50mL) of sodium bicarbonate and hemodialysis for his severe acidosis and oliguric kidney failure.
On hospital day 2-3, our patient’s mentation worsened despite treatment of his sepsis and hypotension and resulting improvement in urine output. He was given a banana bag (dextrose based infusion of multivitamins including thiamine, folic acid and magnesium) and started on tube feeds through a nasogastric tube.
On hospital day 4, his HAGMA, which had previously improved with hemodialysis, recurred.
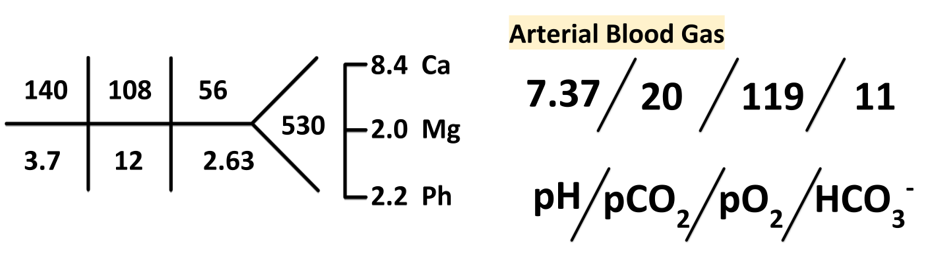
A focused workup for the recurrent HAGMA was pursued, which demonstrated a serum beta hydroxybutyrate = 5.3 mg/dL (normal < 2.8 mg/dL).
E. Final Diagnosis and Management
Our patient’s clinical course suggested two distinct causes of metabolic acidosis occurring in sequence. His initial acidosis was likely due to accumulation of sulfates and phosphates from kidney injury. The later development of hyperglycemia and ketoacidosis suggests diabetic ketoacidosis as the cause for recurrent acidosis.
Let’s discuss the differential for ketoacidosis.
Ketoacidosis is metabolic acidosis due to accumulated ketone bodies. There are three major ketone bodies:
- Acetoacetate (a true ketone and acid)
- β-hydroxybutyrate (an acid formed from breakdown of acetoacetate, but not a true ketone itself)
- Acetone (a ketone, however not acidic)
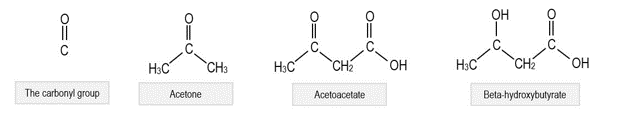
Note that the urine dipstick on admission demonstrated trace ketones, however the urine dipstick only detects acetoacetate. A low or negative ketone test on urine dipstick does not rule out DKA. In our patient, no urine dipstick was tested when he developed DKA a few days into his hospitalization.
How could you have low acetoacetate, but still high beta hydroxybutyrate in ketoacidosis?
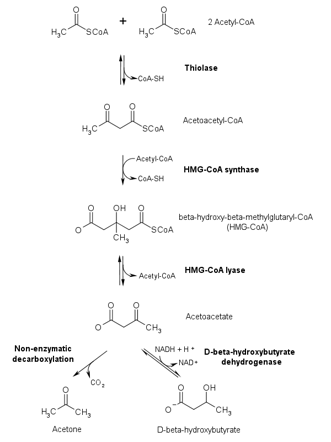
This has to do with how ketone bodies are generated (ketogenesis) and how this process is affected by the type of ketoacidosis present. Recall that ketogenesis starts with acetyl-CoA in the liver and that acetone and beta hydroxybutyrate are both produced from acetoacetate.
His clinical history of hyperglycemia concurrent with development of ketoacidosis in an acutely ill diabetic who started enteral feeding without insulin is consistent with DKA. However given his dementia and failure to thrive, how can we tell if he has starvation ketoacidosis as well? In another situation, the third type of ketoacidosis to consider would be alcoholic ketoacidosis (AKA).
How can we differentiate between DKA, AKA, and starvation ketoacidosis?
Data suggests that the ketone body that predominates (beta hydroxybutyrate versus acetoacetic acid) may help to elucidate. Normally, the amount of beta hydroxybutyrate and acetoacetic acid are about equal, but in AKA and DKA, beta hydroxybutyrate predominates. This is most prominent in AKA because the metabolism of alcohol by alcohol dehydrogenase produces NADH, fueling hepatic conversion of acetoacetic acid to beta hydroxybutyrate. In DKA, the high generation of acetoacetic acid favors the reaction towards the generation of beta hydroxybutyrate. However, high levels of acetoacetic acid lead to a ratio of beta hydroxybutyrate to acetoacetic acid that is not as high as in AKA. In starvation ketoacidosis, on the other hand, the relative lack of insulin from starvation does not typically lead to as profound an acidosis.
How do we treat ketoacidosis?
Insulin is a potent inhibitor of ketogenesis because 1) it inhibits lipolysis by deactivating hormone-sensitive lipase in adipocytes, 2) it activates acetyl-Coa carboxylase, which decreases uptake of fatty acids into hepatic mitochondria where ketogenesis occurs, and 3) it inhibits HMG-CoA synthase, the rate limiting step in ketogenesis. This is the underlying principle for treatment for ketoacidosis of any etiology. In AKA and starvation ketoacidosis, we initiate oral intake or dextrose based infusions and rely on secondary endogenous insulin release to reverse the ketogenesis. In starvation ketoacidosis, we must also monitor closely for electrolyte derangements due to refeeding syndrome. In DKA, insulin is given directly to reverse the ketogenesis until the anion gap is normalized with glucose infusions as needed to mitigate hypoglycemia.
The patient’s hyperglycemia and ketoacidosis was managed by insulin drip, and in the setting of hypoglycemia, he was also given D5 ½ NS. Kidney function continued to improve with supportive care.
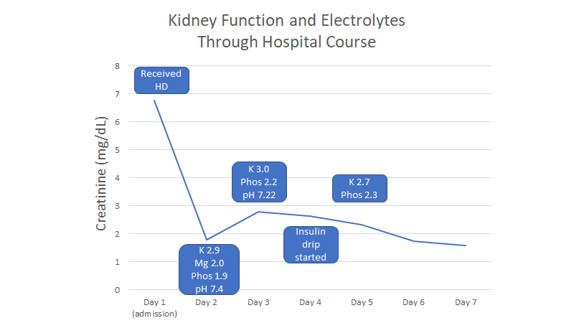
During his hospitalization, our patient also developed hypokalemia, hypomagnesemia, and hypophosphatemia.
What are the etiologies of potassium derangement in DKA?
- The treatment of DKA with insulin drip. Insulin itself shifts potassium into cells.
- Extracellular hypertonicity in DKA can lead to hyperkalemia independent of acidosis through unknown mechanisms, and resolution of hypertonicity would expect to decrease plasma potassium.
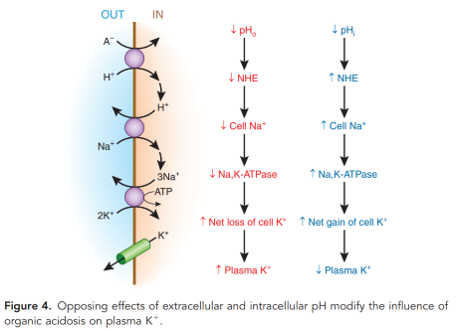
- Acidosis in DKA decreases intracellular potassium uptake by Na+-K+ ATPase via coupling with decreased activity of Na+-H+ exchange. There are other possible transporters that may be involved in coupling movement of H+ and K+, however the effect is thought to be decreased with acidosis due to organic anions (e.g. lactate, ketoacids). With ‘organic acidosis’, transporters move organic anions intracellularly, lowering intracellular pH and promoting Na+-H+ exchange and keeping Na+-K+ ATPase active.
4. The patient had hypomagnesemia. Hypomagnesemia leads to urinary potassium wasting.
In patients with intact renal function, total body potassium deficit tends to occur in DKA due to potassium wasting with glucosuria. However, with improving renal function, it is possible that the patient had urinary K loss due to recovering tubular dysfunction.
F. Take-Home Points
- Urine dipstick detects only acetoacetate, and not beta hydroxybutyrate. Because of this, ketone testing by urine dipstick can miss DKA, and especially AKA.
- DKA, AKA and starvation ketoacidosis may in part be differentiated by the ratio of elevated acetoacetate vs beta hydroxybutyrate. Insulin inhibits lipolysis and ketogenesis and hence is useful in the management of ketoacidosis.
- Hypokalemia during the clinical course of DKA can be due to resolving plasma hypertonicity, resolving organic acidosis, intracellular shift secondary to insulin, osmotic diuresis and recovering tubular function specific to this case.


In my experience there are differing opinions. I prefer to let them eat from the beginning if they can and handle any prandial glucose spikes with additional insulin. It’s cruel to make someone NPO unnecessarily.
Good morning.
I have this doubt:
In a patient with DKA, when is the right time to start oral feeding?
Do we have to wait until the lab resolution of the DKA?