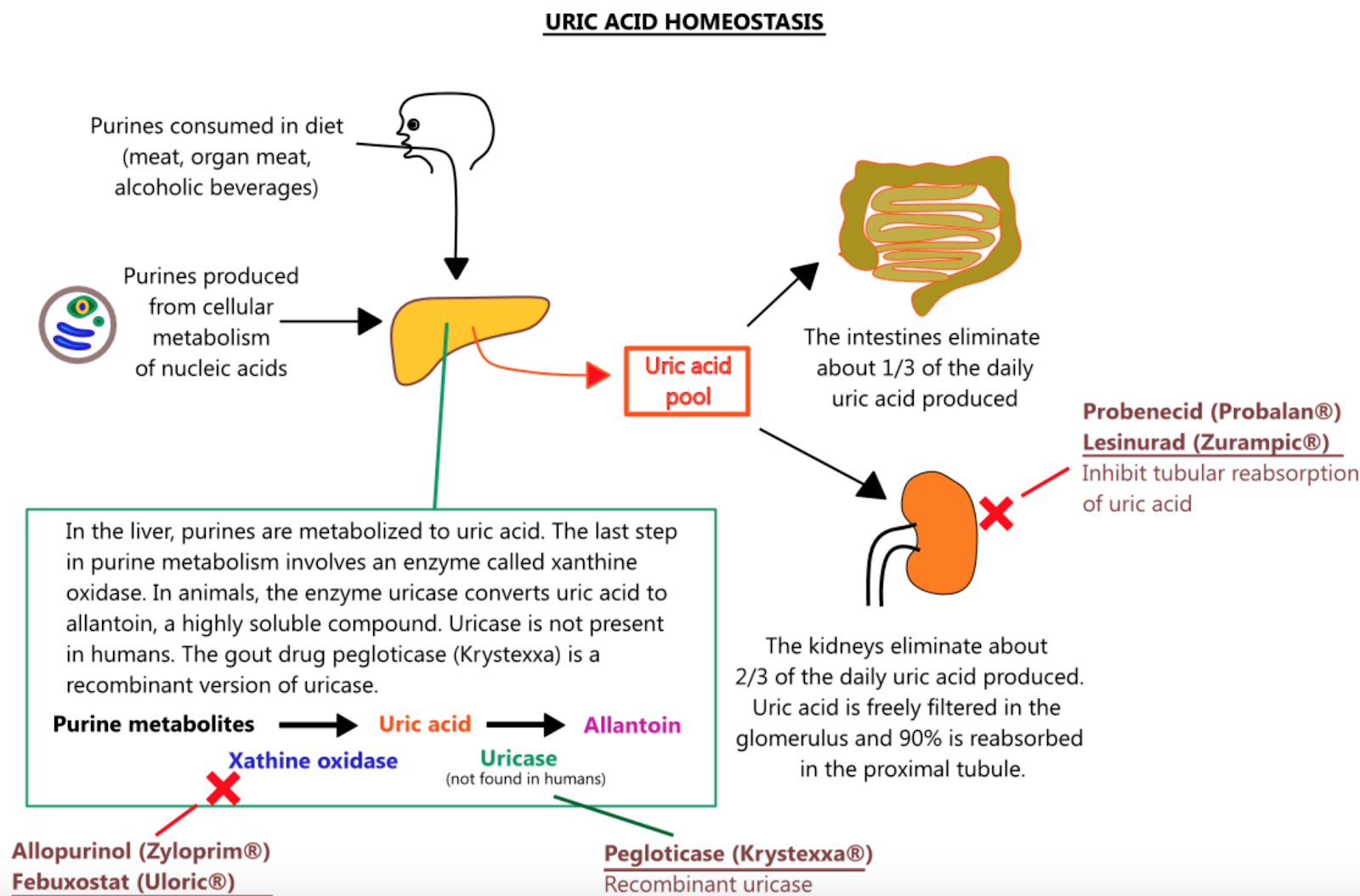
Understanding chronic kidney disease (CKD), and its associated health risks, has been a priority for clinical and basic researchers alike. For years, the role of serum uric acid levels in CKD as a marker or causal factor have been heavily debated (Figure 1). Recently, an association between gout and CKD has been reported, showing a threefold higher hazard of CKD in gout patients over 65 years old, independent of other risk factors. Gout is ultimately caused by the accumulation of monosodium urate crystals and affects 2-3% of people in the US. Despite the unknown mechanism of gout, it is suggested that the effects on CKD may include the activation of inflammatory, proliferative, and other maladaptive pathways by elevated levels of urate.

The first line therapy for gout and hyperuricemia is often allopurinol, a prototypical xanthine oxidase inhibitor, which prevents the conversion of purine metabolites to uric acid. However, the KNOW-CKD and FEATHER studies have demonstrated that allopurinol and febuxostat, another urate lowering therapy, do not elicit renoprotective effects. Though these treatments are a reasonable first option for many patients with gout, understanding cross-talk between uric acid and kidney damage is imperative to develop newer agents which could improve clinical outcomes for patients with CKD (in terms of renoprotection). (Figure 2).
While many investigators have interrogated gout pathogenesis, the laboratories of Tin, Woodward (@goutyprof), and Köttgen have taken a different and exciting approach to teasing out this elusive mechanism by identifying rare functional variants that can potentially serve as molecular targets for reducing elevated serum urate and preventing gout. Current therapies leave patients undertreated and susceptible to recurrent gout flares, making identification of these new targets critical. This multidisciplinary research team employed large scale whole exome sequencing (WES) to identify low-frequency or rare variants with the goal of identifying novel genetic associations that may be able to translate into mechanistic understanding. In WES, all of the exons of protein coding genes, or the exome, are sequenced using highly specific oligonucleotide probes to bind fragmented DNA, which can then be sequenced and subjected to bioinformatic analysis to find rare variants.
Astonishingly, two genes of urate transporters, SLC22A12 and SLC2A9, were identified to have many potentially damaging variants associated with serum urate levels in this large scale WES association study with up to 19,517 European and African-American ancestry. SLC22A12 and SLC2A9 encode urate transporter 1 (URAT1) and glucose transporter 9 (GLUT9), respectively. Both of these proximal tubule transporters serve a critical role in the reabsorption of urate (Figure 3).
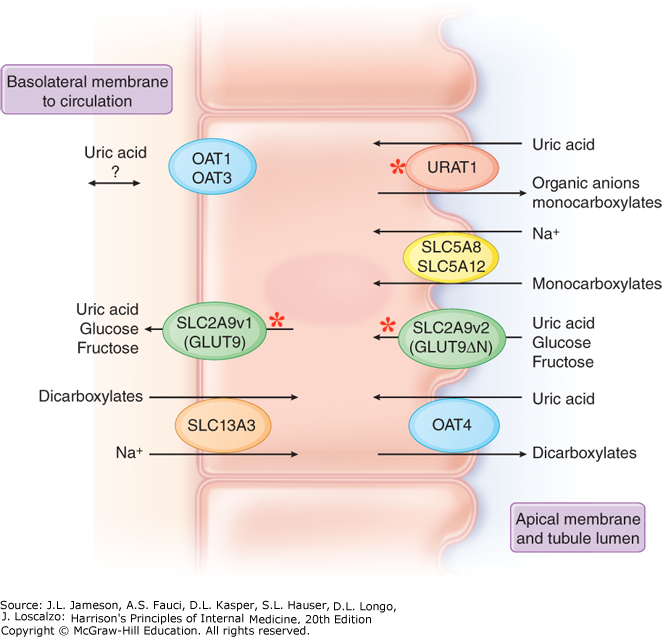
In order to further understand the physiological significance of these variants, the abundance, glycosylation, and transport function of SLC22A12 variants was compared to wild-type URAT1 in several in vitro assays. Each of the variants (R325W, R045C, and T467M) demonstrated decreased abundance, an altered ratio of maturely glycosylated to immature protein, and reduced urate transport in a C-14 radiolabeled urate accumulation assay, consistent with loss of function. By identifying loss of function variants in URAT1, Tin et al. provides potential evidence for the therapeutic value of urate-lowering drugs for hyperuricemia and gout treatment by inhibiting proximal tubule urate uptake and promoting urate excretion.
For SLC2A9, Tin et al. employed a parallel approach to interrogate mechanism by identifying functional domains in GLUT9 where rare variants accumulated, revealing three domain “hotspots.” These domains include the intracellular interface, extracellular interface, and central core region. While it is predicted that the identified mutations may not change structure, alteration of residues in these domains may change the binding and affinity of urate, resulting in consequences in transport capacity. The experimental approach from large-scale WES association study to the mapping of variants to protein function eloquently presented in this paper will be imperative for designing in vivo experiments to further understand urate transport. Ultimately, this will lead to the critical design of new urate-lowering therapies that not only will aid in the reduction of gout burden, but also may improve risk of CKD in this patient population.
Eryn E. Dixon
PhD Candidate
University of Maryland School of Medicine
Department of Physiology

