Our focus on myeloma related disorders continues- view previous posts here.
Pathogenesis
C3GN is a type of glomerulonephritis characterized by dominant C3 deposition resulting from overactivation of the alternative pathway of complement (APC) from acquired or hereditary causes. The most common mechanism of APC overactivation is development of autoantibodies to C3 convertase, called C3 nephritis factor (C3NeF), which stabilize the convertase and lead to pathway overactivation. C3NeF is also commonly present in the related entity dense deposit disease (formerly MPGN type II). Some patients may instead harbor mutations in APC regulatory proteins such as CFH, CFI, MCP/CD46, and CFHR. These mutations also cause complement dysregulation in the setting of atypical hemolytic uremic syndrome.
Finally, monoclonal immunoglobulins in the setting of a hematopoietic neoplasm (most commonly a plasma cell dycrasia) can activate the APC. Overactivation of the APC leads to deposition of C3 in the glomeruli, which can lead to an inflammatory reaction and resulting glomerular injury. In many patients, the development of C3GN is preceded by a complement-amplifying condition such as an infection. Interestingly, it has been noted that some patients with post-infectious glomerulonephritis show persistent C3 deposition instead of fully recovering, and this so-called “resolving/atypical” post-infectious glomerulonephritis is thought to represent the development of C3GN in patients with APC abnormalities.
Clinical presentation
Patients with C3GN present at different ages depending on the underlying pathogenesis. C3GN resulting from abnormalities of the APC generally presents in childhood or young adulthood. In comparison, C3GN resulting from an underlying monoclonal gammopathy typically presents in older adulthood. The clinical presentation is variable and often reflects the specific pathologic pattern of injury present on biopsy. Hematuria and at least mild proteinuria are present in the majority of cases. History of infection is not uncommon. Cases demonstrating membranoproliferative features on biopsy are often accompanied by more severe proteinuria. The presence of chronic renal failure suggests the presence of irreversible scarring on biopsy. Complement C3 levels are often decreased, but a normal C3 level does not exclude C3GN. C4 levels are usually normal. C3NeF is present in approximately 50% of cases.
Genetic testing may uncover mutations in components of the alternative pathway of complement such as CFH, CFI, MCP/CD46, and CFHR. In older adults, a history of hematopoietic malignancy may already be established, but the diagnosis of C3GN in an older patient without a known history of monoclonal gammopathy should prompt a monoclonal workup by protein electrophoresis and/or free light chain assay. Treatment in these patients should center around addressing the underlying monoclonal gammopathy. For complement-driven C3GN, a variety of treatments have been attempted but inhibition of complement with anti-C5 therapy has been effective in a subset of patients, and plasmapheresis has been used for autoantibody-mediated disease. C3GN progresses to end-stage renal disease in up to 50% of cases, and frequently recurs in the allograft.
Light microscopy
A spectrum of findings can be present by light microscopy. Most cases show a mesangioproliferative pattern of injury characterized by hypercellularity in the mesangium. A subset of cases will additionally show endocapillary hypercellularity, frequently including neutrophils within the glomerular capillaries similar to the exudative changes seen in post-infectious glomerulonephritis. Cases with endocapillary hypercellularity may also be associated with crescentic injury. An additional subset of patients show a membranoproliferative (MPGN) pattern of injury, including mesangial hypercellularity and duplication of the glomerular basement membranes with or without endocapillary hypercellularity and crescents. The tubulointerstitium will show variable degrees of fibrosis and atrophy. Thrombotic microangiopathy has rarely been observed in the setting of C3GN, likely related to the common pathophysiology of APC dysregulation.
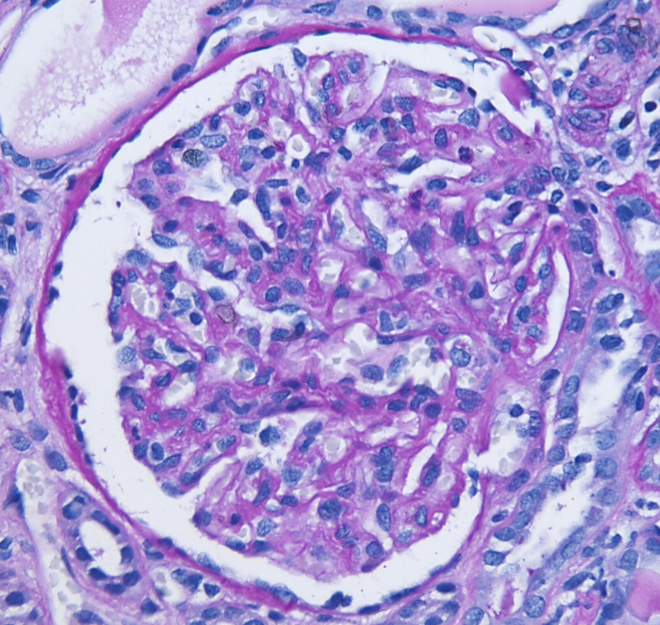
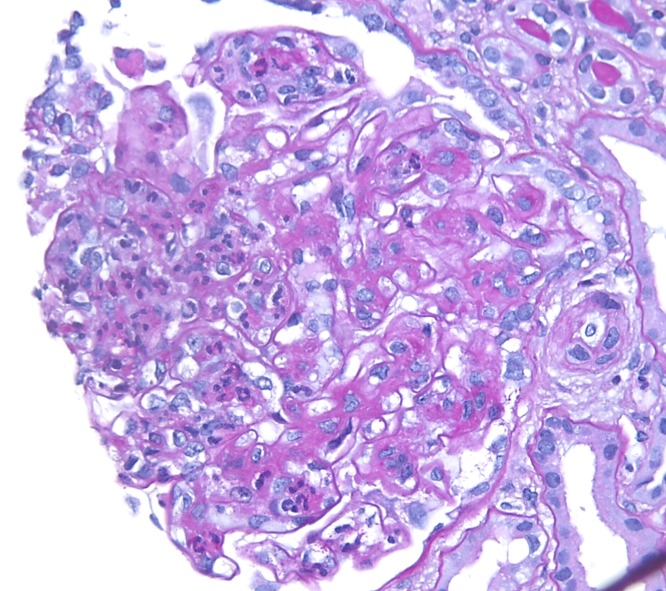
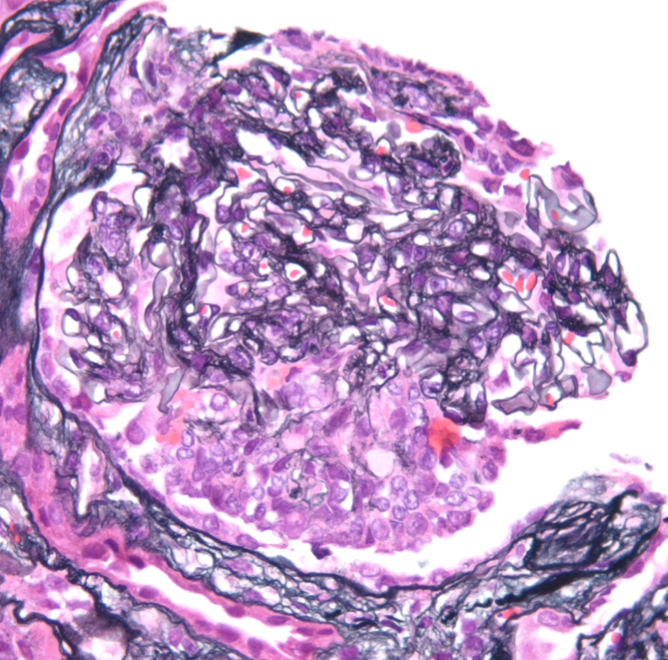
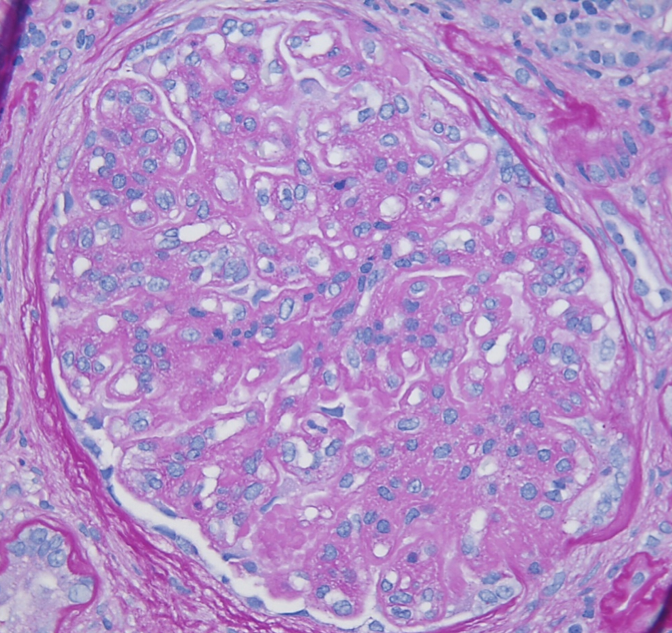
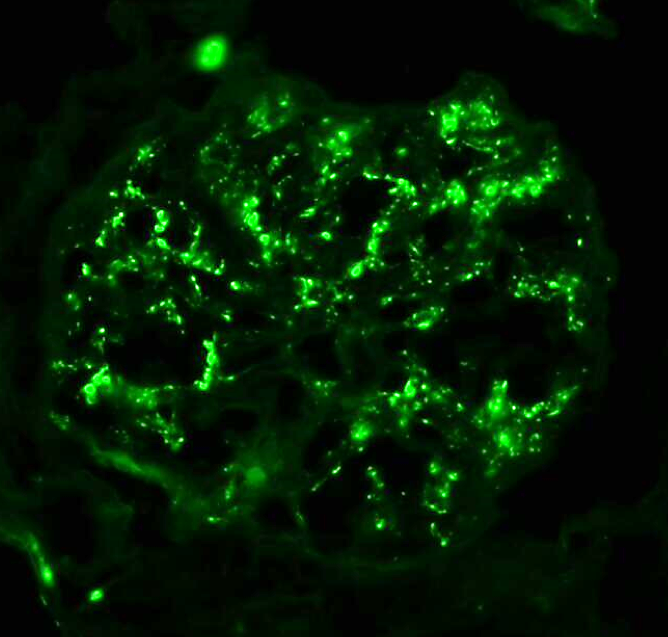
Immunofluorescence microscopy
C3GN is defined by the presence of dominant C3 deposition in the glomeruli. In most cases, C3 is strongly present with little or minimal immunoglobulin deposition. However, immunoglobulin deposition is allowable as long as C3 deposition is at least 2 levels of intensity stronger (ex: C3: 3+, IgG: 1+). The pattern of granular C3 deposition typically correlates with the light microscopic findings. Mesangial-only C3 deposition is typically seen in mesangioproliferative C3GN, whereas mesangial plus capillary wall C3 deposition is usually associated with endocapillary proliferative or membranoproliferative forms of C3GN by light microscopy. C4d is typically negative or shows minimal deposition, which may be useful in distinguishing C3GN and post-infectious glomerulonephritis in some instances.
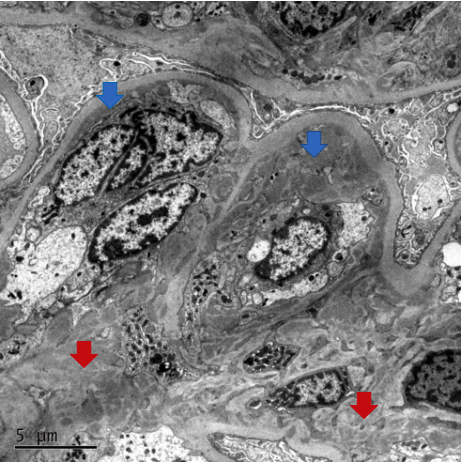
Electron microscopy
Electron microscopy in C3GN demonstrates the presence of amorphous electron-dense deposits in a pattern that typically correlates with the light microscopic findings. Mesangial-only deposits are typically seen in mesangioproliferative C3GN. Additional subendothelial deposits are commonly present cases showing endocapillary hypercellularity or MPGN patterns. Occasional subepithelial or intramembranous deposits may also be present. While the deposits of C3GN are electron dense, the absence of confluent hyperdense deposits allows distinction from dense deposit disease.
Differential Diagnosis
- Post-infectious glomerulonephritis – Typically shows an exudative pattern by LM, usually IgG, C4d, and C3 deposits by IF, and subepithelial “hump-like” deposits on EM.
- Dense deposit disease – Hyperdense “sausage-like” deposits by electron microscopy.
- Membranoproliferative glomerulonephritis with immune complex deposition and Infection-related glomerulonephritis – Usually Ig, C4d, and C3 deposition, although infection-related cases may show dominant C3.
- Membranous-like glomerulonephritis with masked IgG-kappa deposits – Routine IF shows C3 deposition without significant Ig deposition, but IF on paraffin tissue digested with pronase demonstrates monoclonal IgG-kappa staining. EM shows subepithelial deposits.
Alexander Gallan, MD
Assistant Professor of Pathology, The Medical College of Wisconsin

