Welcome to the 9th case of the Skeleton Key Group, a team of twenty -odd nephrology fellows who work together to build a monthly education package for Renal Fellow Network. The cases are actual cases (without patient identifying information) that intrigued the treating fellow.
Written by: Kartik Kalra
Visual Abstract: Sophia Ambruso
A. The Stem
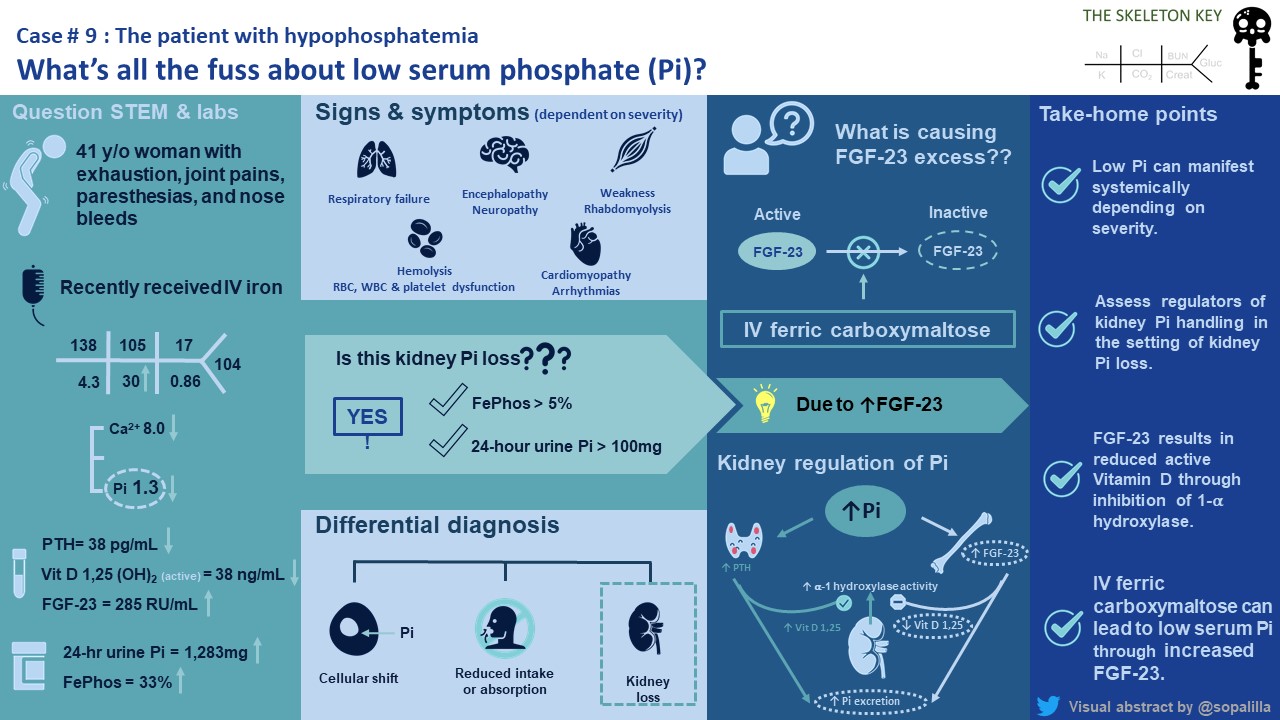
A 41-year-old female with a history of systemic lupus erythematosus (SLE), Raynaud’s phenomenon, migraines, and depression is referred to the nephrology clinic for refractory hypophosphatemia. Two weeks ago, she saw her rheumatologist with complaints of malaise, nausea, and constipation over the past two to three months. Due to a low serum phosphorus (Pi) of 1.1 mg/dL, she was referred to the emergency department without concern for active lupus nephritis. She was started on oral phosphorus supplementation and followed up after 5 days. Labs showed persistent hypophosphatemia of 1.3 mg/dL, and she was urgently referred to nephrology.
In the office, she complained of exhaustion, new epistaxis, joint pains, “pins and needles” in hands and feet without frank weakness, and nausea. She admits to starting an intravenous (IV) iron infusion for iron deficiency in the past few months with no other changes in medications. Review of systems was otherwise unremarkable.
Vitals: BP 113/77 mm Hg | Pulse 79 bpm | Temp 98.7 °F (37.1 °C) (Oral) | Resp 20 per minute | BMI 26.46 kg/m²
CV: RRR, normal S1/S2
Pulm: no conversational dyspnea, no increased work of breathing
Neuro: No focal neurologic deficits, reflexes 2+, 4/5 muscle strength in bilateral upper and lower extremities
Remainder of exam was within normal limits
B. The Labs
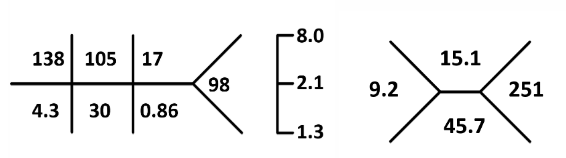
pH 7.41 Albumin 3.8 iCal 1.19
C. Differential Diagnosis
This patient has many symptoms of hypophosphatemia which include fatigue, numbness, paresthesia and epistaxis (Table 1, Fig 1). Her creatine kinase (CK) level was 66 units/L and she showed no evidence of rhabdomyolysis or myopathy.

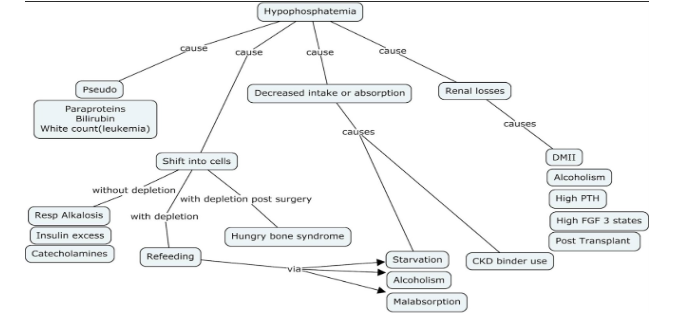
When thinking about hypophosphatemia it is useful to divide the etiologies (Fig 2). Our patient did not appear to have any evidence of transcellular shifting or decreased intake or absorption. She was not a diabetic nor had signs of decreased oral intake from starvation or alcoholism.
Although our patient did have an alkalosis on initial labs. Generally acute respiratory alkalosis leads to extracellular CO2 diffusion which in turn leads to a rise in intracellular pH. The increased intracellular pH increases glycolysis, a process where phosphates are needed to produce adenosine triphosphates (ATP). The requirement for cellular phosphate uptake leads to hypophosphatemia. Chronic respiratory alkalosis is associated with hyperphosphatemia due to kidney resistance to parathyroid hormone (PTH).
Acute metabolic alkalosis does not cause hypophosphatemia because bicarbonate cannot freely cross cell membranes to cause a rapid rise in intracellular pH. Our patient did not have a respiratory alkalosis and her pH was normal.
Next, we wanted to assess kidney handling of phosphorus.
Is the kidney wasting phosphorus?
Let’s do some math: Fractional Excretion of Phosphorus (FePi):

In the case of this patient:
FePi: [ UPhos x SCr ] / [ SPhos x UCr ] x 100 = 0.329 x 100% = 32.9%
FePi: 32.9 %
Kidney phosphate wasting is suggested when the 24 hour urine phosphate is > 100 mg or if FePi > 5% (normal 15-20%) in the setting of hypophosphatemia.
D. More Data
Based on the schema (Fig 3) now we know our patient is having kidney phosphorus wasting, but why?
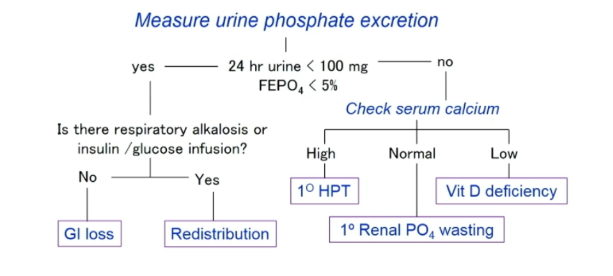
Additional labs were obtained (Table 2). She was not on any diuretics, glucocorticoid, or mineralocorticoid therapy. She had no glucosuria to suggest a Fanconi syndrome, and her free light chain ratio was unremarkable when looking for paraproteinemia related tubular abnormality.
| Intact PTH | 31 pg/mL (normal range 18.4-80.1 pg/mL) |
| PTH related peptide | <2.5 pmol/L |
| 25 OH Vitamin D | 51 ng/ml |
| 1,25 OH Vitamin D | <8 pg/ml |
| Intact Fibroblast Growth Factor 23 (FGF23) | 285 RU/mL (normal ≤180 RU/mL) |
| SPEP | Normal protein electrophoresis, no monoclonal protein identified |
| Serum Immunofixation | Normal pattern. IgG, IgA, IgM all within normal limits. |
| Serum Free Light Chains | NormalK/L ratio = 11.9/10.2 = 11.17 (normal 0.26=1.65) |
E. Final Diagnosis & Management
An astute review of new medications started three months ago indicates she was started on IV ferric carboxymaltose therapy for her iron deficiency anemia. IV ferric carboxymaltose therapy (Injectafer) can lead to increased FGF 23 levels and hypophosphatemia.
Symptomatic hypophosphatemia requiring clinical intervention has been reported in patients at risk of low serum phosphate in the postmarketing setting.
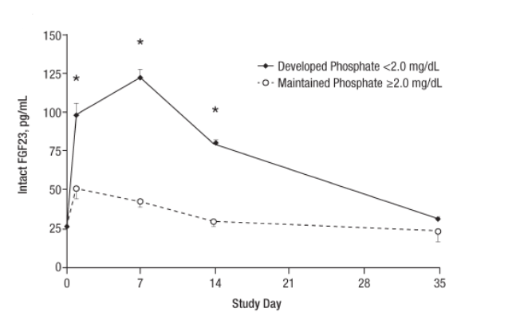
The exact mechanism is unclear, but it is thought to be secondary to increased FGF 23 levels and reduced FGF 23 degradation. A proposed mechanism (Fig 4) looks at the balance of iron and FGF 23 levels. In iron deficiency, there is increased FGF 23 transcription, but also deactivation of active FGF 23. When we correct iron deficiency, FGF 23 transcription normalizes. But other iron infusions (specifically ferric carboxymaltose) can also inhibit FG23 degradation resulting in increased FGF 23 levels, such as seen in our patient. The hypophosphatemia from IV iron infusions peaks around 7 days and may be prolonged lasting over months after infusions are complete (Fig 5).
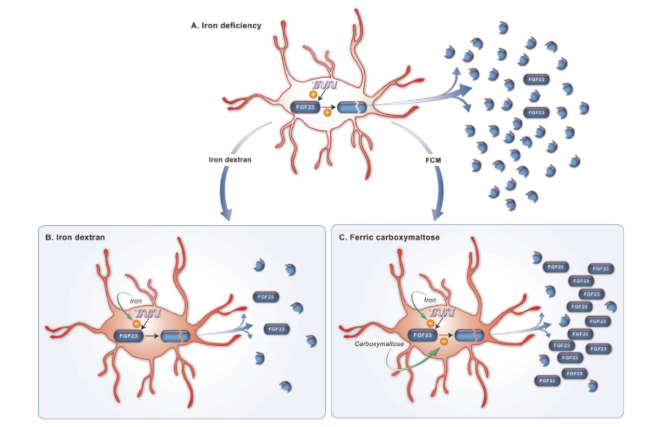
Fig 4. Proposed Mechanism of IV Ferric Carboxymaltose leading to Hypophosphatemia
Interestingly our patient also had a low 1,25 OH Vitamin D level. Can you guess why? Let’s review quickly how kidney phosphorus handling is influenced by FGF 23, PTH, and Vitamin D.
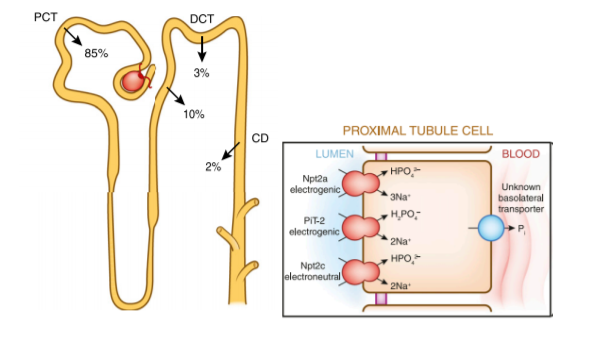
PTH and FGF 23 are phosphaturic via downregulation of the expression of NaPi 2a and 2c. FGF 23 also reduces 1,25OH Vitamin D levels through inhibition of 1∝ hydroxylase in the proximal tubule, which is one of the main sites of vitamin D hydroxylation (Fig 7a and 7b).
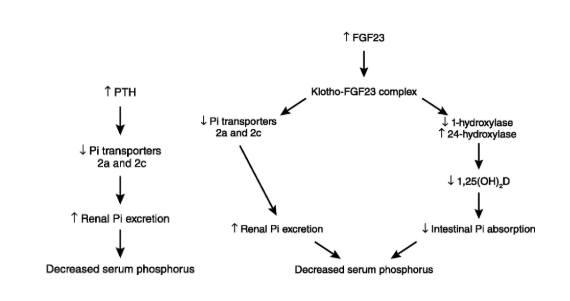
Fig 7. PTH and FGF 23 regulation of kidney handling of phosphorus (Felsenfeld AJKD 2012)
Management: depends on if the patient’s presentation is acute or chronic.
Acute hypophosphatemia can be life threatening causing arrhythmias, specifically when severe (<1 mg/dL) requiring IV phosphorus replacement.
Our patient would be categorized as chronic. Her severity (Table 1) is moderate, and moderate chronic hypophosphatemia can typically be managed with oral phosphorus repletion and active vitamin D therapy.
She was started on oral phosphorus repletion, calcitriol, and dipyridamole. Her phosphorus level, FGF23, and 1,25OH Vitamin D levels normalized 12 months from her last IV ferric carboxymaltose infusion with no further requirement for supplementation (Fig 8). She was further advised to avoid all further IV ferric carboxymaltose infusions.
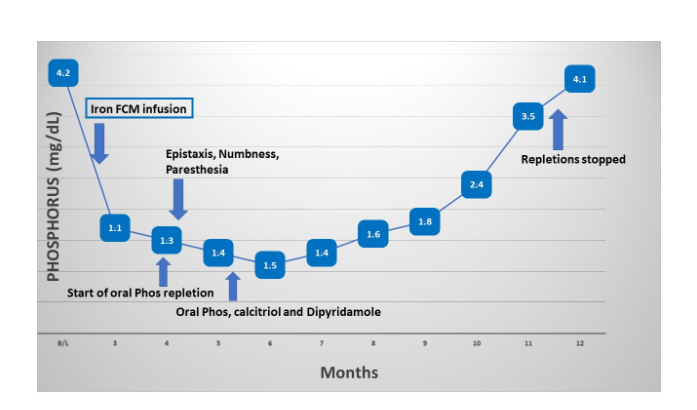
Fig 8. Patient’s Phosphorus Levels over Time
TAKE HOME POINTS
- Symptoms of hypophosphatemia can include muscle weakness, and severe hypophosphatemia can lead to respiratory depression and cardiac arrhythmias.
- Clinicians should be aware that IV ferric carboxymaltose can lead to hypophosphatemia through FGF23 and can persist months after infusions are complete.
- Assess regulators of kidney handling such as FGF 23, Vitamin D, and PTH when looking at kidney phosphorus wasting
- FGF23 results in decreased 1a hydroxylation, affecting levels of 1,25 OH VItamin D and PCT Pi reabsorption


That is a great differential Satish! Although pretty rare. Both present with nonspecific symptoms such as bone pains and muscle weakness. Both also cause renal phosphate wasting, increased FGF23, and low or low-normal 1,25 (OH)2D. Usually though, these patients will present with focal symptoms (bone pains) , or symptoms like weight loss, fatigue, etc. or palpable or visible bone abnormalities (pubmed.ncbi.nlm.nih.gov/29021995/). In this case we wait and see– and if no change in a few months we can proceed with imaging (MRI would be preferred over pet/CT).
Great question Olaf! Hypophosphatemia has been reported mostly in ferric carboxymaltose and some in iron sucrose (Venofer). Mechanism is postulated to be due to the inhibition of FG23 degradation resulting in increased FGF 23 levels and subsequent phosphaturia. No reports found for ferric gluconate (Ferrlecit) so far.. A nice systematic review of hypophosphatemia associated with IV iron therapies is found here: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7152545/
Here, hypophosphatemia rates ranged from 0% to 92% for ferric carboxymaltose and 0% to 40% for iron sucrose,
Does it occur with fellecit and venofer as well??
Congratulations for the post
What about Tumor Induced Osteomalacia. High FGF 23 leading to low P. Should we do imaging for phosphaturic mesenchymal tumor , with Ga dotatate PET/CT before confirming that it is due to FCM. Tumor may remain hidden for long and even in proven cases treatment might be suppplements short of resection.
Dipyridamole decrease renal phosphate wasting in patient with with low renal threshold for phosphate.
Hi Dave, great question that has been asked by a few people here. Adenosine is taken up by tubular cells and this produces phosphaturia, under the control of PTH. This effect is blunted by dipyridamole as it blocks the transport of adenosine from the lumen into the tubular cells. This effect was demonstrated nicely in possums in 1992 here (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC329939/) and in humans in 1994 here (https://www-nejm-org.proxy.medlib.uits.iu.edu/doi/full/10.1056/NEJM199407073310122).
Very informative post!
Why is been given dipiridamole?
Nice one!! Hope to learn more about P metabolism
What’s d reason for giving dipyridamole in the management?
Interesting… you advise measuring FePi on 24h collection. Plasma Pi concentration varies (a lot!) all over the place in 24h so, I really think spot urine assessment (TmPi In this case, K-to-Cr ratio in hypoK) is more accurate. Really nice case report btw!