Eryn Dixon
Postdoctoral Fellow
Washington University School of Medicine in St. Louis
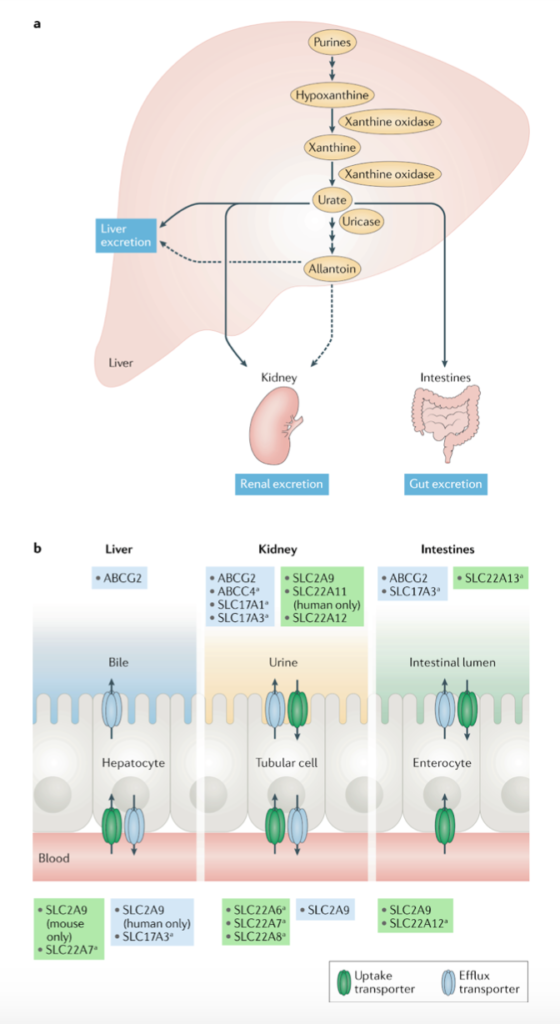
Uric acid is the final metabolic byproduct of purine nucleotide breakdown (Figure 1a) and its accumulation causes hyperuricemia, which is highly prevalent and heritable. The major consequence of this pathological elevation of serum urate, which affects 43 million Americans (Woodward et al, 2015), is gout, but it can also be accompanied by kidney stones. Both the gastrointestinal tract and the kidneys are responsible for the excretion of urate, the salt form of uric acid, in humans, but the specific mechanisms are unknown. Variants in three transporters, including Glut9, URAT1, and ABCG2, that have an affinity for urate have been identified as major contributors to hyperuricemia due to their prevalent effects on serum urate variability (Figure 1b). One of these transporters, ATP-binding cassette subfamily G member 2 (ABCG2), encoded by the highest risk gout gene ABCG2, is a multidrug resistance protein andbelongs to the ATP-binding cassette (ABC) family. There has been much discrepancy in both in vivo and human studies in regard to the expression, localization, and function of ABCG2.
In human disease, ABCG2 has a common single nucleotide polymorphism (SNP) associated with increased serum urate levels. This SNP results in a missense variation in the ABCG2 protein (Q141K) (Woodward et al, 2009). With associated comorbidities of hypertension, obesity, and chronic kidney disease, it is more important than ever to map the molecular mechanisms of hyperuricemia and define the role of ABCG2 in the gastrointestinal tract and kidney. In order to aid in the understanding of the mechanisms of hyperuricemia as well as the functional role of ABCG2 in physiology and disease, Hoque et al set out to develop a mouse model that would recapitulate the increased serum urate that characterizes human disease specific to the disease variant Q141K (Hoque et al, 2020). Previously, human studies have demonstrated increases, decreases, and no response to Q141K in renal fractional excretion of urate.
The mouse model uses a CRISPR knock-in of the orthologous Q140K Abcg2 and exhibits a significant increase in serum urate (SU) in mice homozygous for Q140K (+/+). Intriguingly, this increase in SU was specific to males, while females showed no such SU responses; a finding that mimicked the sex differences of human Q141K variant disease (Köttgen et al, 2013; Tin et al, 2019). Importantly, these changes in the Q140K+/+ mice were independent of any alterations in diet or uricase (an enzyme in mice that breaks down uric acid) function. Additionally, these Q140K+/+ male mice exhibited significantly decreased fractional excretion of uric acid (FEUA), which supports the dependence of SU on FEUA.
Next, Hoque and colleagues sought to define the potential changes of abundance and localization of ABCG2 in the Q140K+/+ mice, compared to controls (wild-type C57BL6J). ABCG2 in the Q140K+/+ mice did not vary in mRNA expression. Again in male Q140K+/+ mice, there was an observed decrease in total abundance. In both the mouse model and human kidney tissue, ABCG2 was confirmed to be in the S2-S3 region of the proximal tubule. In the Q140K+/+ mouse model, there was a change in immunofluorescent signal of ABCG2 at the brush border of the proximal tubules, falling in line with the observed decreases in ABCG2 protein. While this paper made great strides in characterizing ABCG2 in the kidney, it expanded its scope to integrate the changes in intestinal urate secretion in the Q140K+/+ mice. Again, ABCG2 protein levels in the intestines were reduced in the Q140K+/+ male mice, even more dramatically than the protein loss observed in the kidney. Emphasis of intestinal loss of ABCG2 in the mouse model was supported through intestinal loop experiments, which demonstrated Q140K+/+ male mice had decreased basolateral to luminal urate flux. This result and other intestinal loop experiments with luminal ABCG2 inhibitor topiroxostat lead Hoque et al to conclude that changes in urate secretion in the intestine may ultimately drive hyperuricemia.
All packed into one paper, the introduction of the Q140K mouse model for hyperuricemia, in conjunction with a human interventional trial that used purine challenges to evaluate the effect of ABCG2 variants on renal excretion, reinforced the relationship between the kidney and the intestines in urate handling (Hoque et al, 2020). This multifaceted approach to understanding these disease mechanisms put forth an interesting model where total urate excretion is driven by the kidney, until a maximum renal excretion level is reached (Figure 2). Once this happens, extrarenal excretion is required to maintain homeostasis. However, when there is a mutation in ABCG2 and the intestines are then unable to fulfill their role in urate excretion, there is resulting hyperuricemia. While there is still much to discover, Hoque et al created an important new system and powerful approach to continue the understanding of hyperuricemia that will lend to the elucidation of its pathophysiology.

Reviewed by: Elinor Mannon, Kelly Hyndman, PhD, Matthew Sparks, MD, FASN, FAHA
If you enjoyed reading this post and are interested in learning more about hyperuricemia and possible clinical presentations and treatment, consider checking out these resources on RFN:
Tweetorials: Understanding Uric Acid, What happened to uricase?