Thrombotic microangiopathy (TMA) is characterized by microvascular endothelial injury and thrombosis, presenting both clinically and histopathologically in two main forms; acute and chronic TMA.
Etiology and Clinical Presentation
Table 1: Etiologies of TMA classified according to clinical presentation
| Typical Hemolytic Syndrome (HUS) | Atypical HUS (& Forms of TMA; Clinical Overlap With TTP) | Thrombotic Thrombocytopenic Purpura (TTP) |
| Enteropathogenic infections (Escherichia coli, Shigella dysenteriae) | Nonenteric infections (e.g., Streptococcus pneumoniae, HIV, Mycoplasma pneumoniae, Coxsackie A & B) | Familial (Mutations in gene encoding ADAMTS13, a vWF cleaving protease; large vWF multimers cause increased platelet aggregation) |
| Genetic defects in complement activation & regulation (mutations in genes CFH, MCP (CD46), CFI, CFB, C3 | Autoimmune (Autoantibodies against ADAMTS13) | |
| Genetic defects in cobalamin metabolism & coagulation pathway | Drugs (Ticlopidine, Clopidogrel) | |
| Drugs (cyclosporine, tacrolimus, sirolimus, anti-VEGF therapy, mitomycin-C, cisplatin, quinine, vaccines) | Hereditary (Homozygous or compound heterozygous ADAMTS13 gene mutations) | |
| Bone marrow transplantation | ||
| Radiation | ||
| Pregnancy & post-partum | ||
| Systemic sclerosis | ||
| Autoimmune (antiphospholipid antibody syndrome, SLE, anti-factor H autoantibodies) | ||
| Malignant hypertension | ||
| Antibody-mediated rejection in transplantation | ||
| Neoplasms such as adenocarcinomas (especially mucin producing) &, rarely, myeloproliferative neoplasms, leukemia & lymphoma | ||
| Idiopathic |
Clinical presentation varies in HUS and TTP; and also based on etiology. Shiga toxin-associated HUS typically affects children less than 5 years of age.
Patients with DGKE, CFH, THBD mutations and Cobalamin C disease present during infancy. Patients with anti-factor H antibodies mostly present 9-13 years of age.
Hereditary TTP typically presents in children; but environmental triggers may precipitate disease in adults. Acquired ADAMTS13 deficiency-mediated TTP occurs mostly in adults (18-50 years); and is seen more often in African Americans and females.
Histopathology
Acute TMA
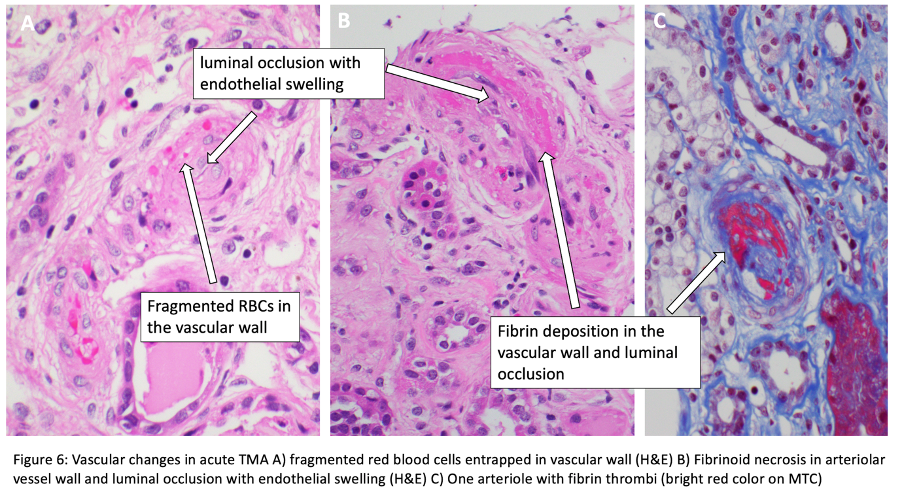
Glomeruli show bloodless appearance, apparent thickening of capillary wall due to subendothelial expansion, mesangiolysis, endothelial swelling, thrombi in the hilum or glomerular capillaries. Nuclear debris present but there is no significant inflammation. Some initial data suggested HUS thrombi are fibrin rich, and TTP thrombi are platelet rich (it is not a reliable feature and not possible to differentiate histologically).
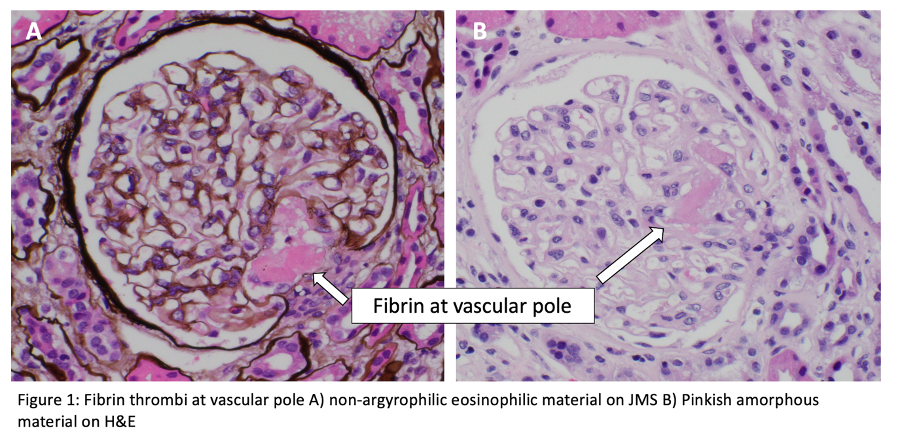
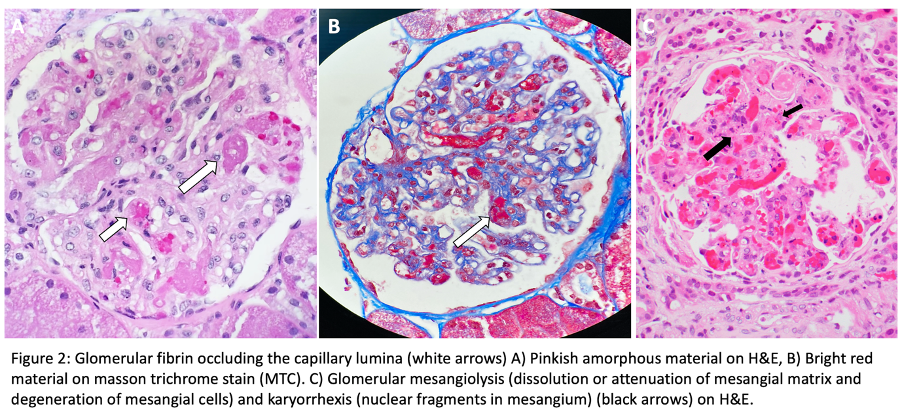
Fibrinoid necrosis may be present. Rare crescents seen in occasional cases.
Fragmented RBCs can be seen entrapped in thrombi, subendothelium, and mesangium. In presence of arteriolar changes, downstream glomeruli demonstrate ischemic collapse of capillary tufts. Segmental sclerosis with collapsing features may be observed.
Tubulointerstitium shows acute tubular injury characterized by simplified epithelium and nuclear reactive atypia. Interstitial edema and cortical necrosis may present in severe cases.
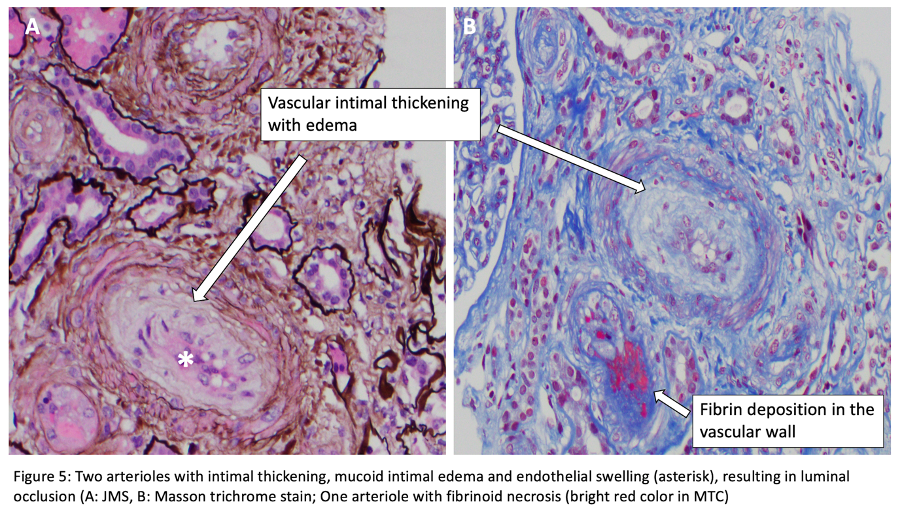
Vessels: Arterioles near vascular pole are affected frequently. There are endothelial swelling and luminal occlusion, intimal mucoid edema with entrapped schistocytes, and thrombi. Fibrinoid necrosis may be present without evidence of inflammatory infiltrate.
Interlobular and arcuate arteries show similar changes.
Chronic TMA
Changes may be seen within days of acute onset of TMA
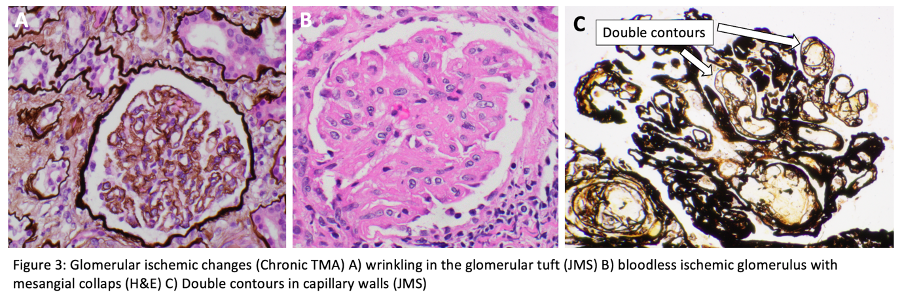
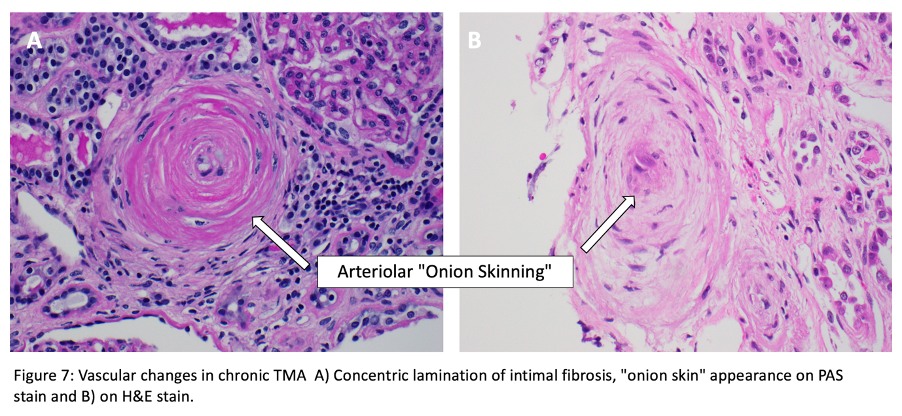
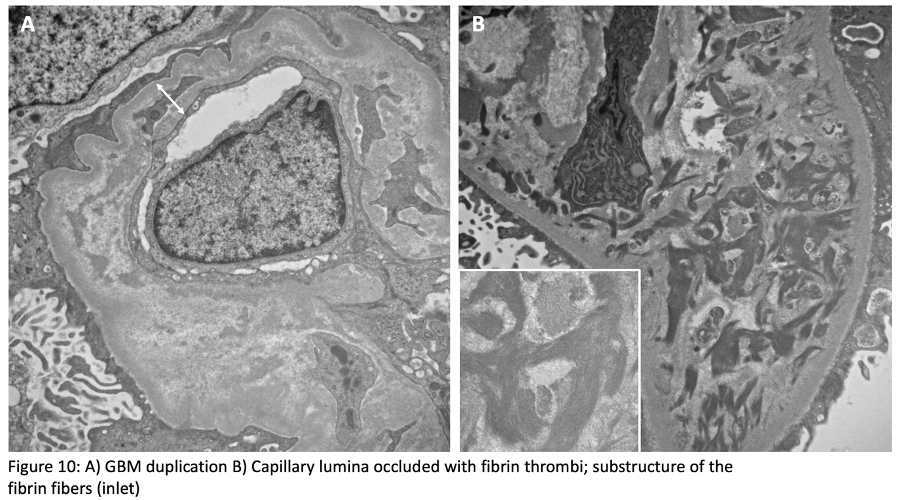
Glomeruli show GBM duplication and double contours (due to endothelial cell injury), mesangial expansion with features of mesangiolysis, variable focal segmental and global glomerulosclerosis.

Tubulointerstitium: There are mostly tubular atrophy and interstitial fibrosis.
Vessels (Arterioles and small arteries): There are intimal fibrosis with narrowed lumina and concentric lamination of intimal fibrosis which causes “onion skin” appearance. Re-canalized thrombi may be identified.
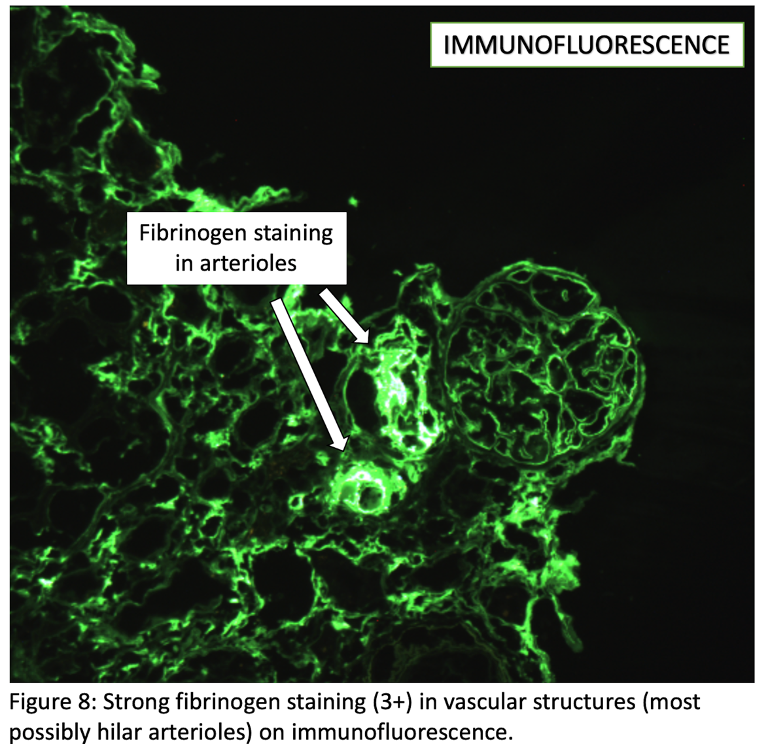
Immunofluorescence Microscopy
Thrombi in glomeruli and vessels stain for fibrinogen. Also, there may be nonspecific entrapment of C3 and IgM in glomeruli.
No immune complex deposition is expected unless etiology is associated with connective tissue disorders such as SLE.
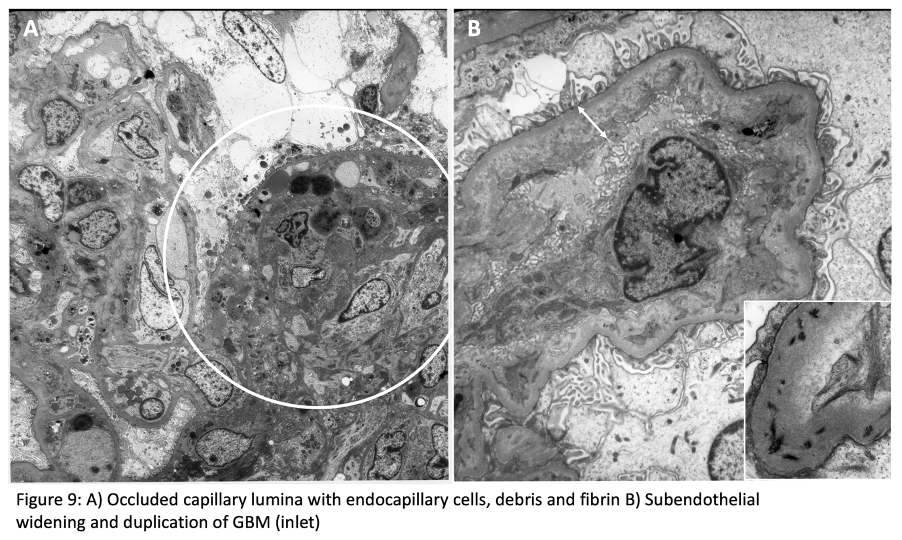
Electron Microscopy
Acute TMA
Capillary lumina shows endothelial swelling and loss of fenestrations. There is expansion of subendothelial space by electron-lucent material. There are entrapped schistocytes and fibrin tactoids in subendothelium. Mesangial matrix is also lucent (mesangiolysis). Podocyte foot process effacement is common. Thrombi show mixture of platelets, fibrin, and erythrocytes. Myocyte degeneration may be seen in affected arterioles.
Chronic TMA
There is increased matrix in mesangium and subendothelium. Depolymerized fibrin may be identified. Duplication of glomerular basement membranes and mesangial cell interposition are frequent. Podocyte foot processes are often effaced.
Diagnostic Pearls
- Isolated involvement of glomeruli is associated with better prognosis than when arterioles also affected
- Cortical necrosis is a poor prognostic factor
- Etiology cannot be distinguished based on biopsy findings
- Anemia, thrombocytopenia, and schistocytes may be absent in mild disease
- Latency period between exposure to radiation and TMA can be very long
- Biopsy with TMA due to malignant hypertension often shows hypertensive arteriopathy
- Significant thrombosis in absence of TMA or arterial remodeling should raise suspicion for antiphospholipid antibody syndrome
Meral Uner, M.D.
Kidney Pathology Fellow
Johns Hopkins Hospital
Instructor/ Staff nephropathologist
Hacettepe University Pathology Department
Ankara, Turkey

Thank you, Excellent
Very good presentation!