Welcome to the 19th case of the Skeleton Key Group, a team of 50-odd nephrology fellows who work together to build a monthly education package for the Renal Fellow Network. The cases are actual cases (without patient identifying information) that intrigued the treating fellow.
Written by: Krithika Mohan, MD
Visual Abstract & Infographics: Mythri Shankar, MD, Sharanya Ramesh, MD, Missy Hanna,MD
A. The Stem
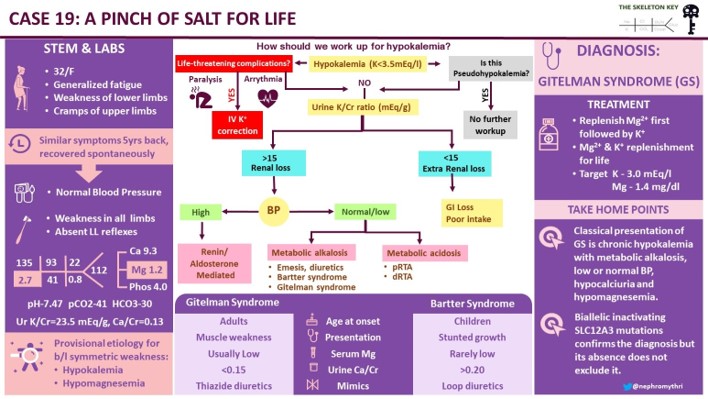
A 32-year-old woman, presented to the primary care clinic with bilateral lower extremity weakness for 2 weeks and difficulty walking for 3 days. She reported occasional bilateral upper extremity muscle cramping. Because of severe fatigue, she had trouble performing her routine daily activities. She had no vomiting, loose stools, constipation, excessive thirst, excessive salt craving, increased urination, paresthesias, or arthralgias. She denied using any medications before the onset of symptoms. She had one episode of weakness of both lower limbs 5 years ago, which improved spontaneously. She did not seek medical treatment, nor did she take any over-the-counter medications. She has no family history of kidney disease.
Vitals:
Heart rate: 80 bpm
Blood Pressure: 106/70 mm Hg
Respiratory rate: 18/min
Oxygen saturation: 100% on room air
Temperature: 98 oF
Focused Physical examination:
General: Alert, oriented
Heart/ Lungs/ Abdomen- Normal
Nervous system: Motor power 3/5 in bilateral lower limbs and 4/5 in upper limbs.
Bilateral ankle and knee jerk reflexes were absent and bilateral plantar were flexor.
Higher mental functions, speech, cranial nerves, sensory examination, and cerebellar functions were normal.
B. The Labs
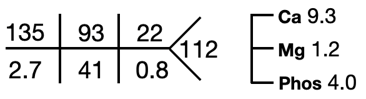
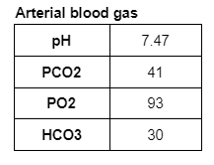
Ultrasound of the abdomen showed normal sized kidneys with normal echogenicity, and no evidence of renal calculi.
Provisional etiology for recurrent, bilateral, symmetrical weakness
- Hypokalemia
- Hypomagnesemia
- Thyrotoxicosis
- Hypokalemic periodic paralysis
- Myopathies
C. Evaluation of Hypokalemia
Step 1 While evaluating hypokalemia the first step would be to look for potential life-threatening complications indicating urgent treatment with IV potassium, such as cardiac arrhythmias or paralysis.
Step 2 The next step is to evaluate the cause of hypokalemia. Our patient’s urine labs showed
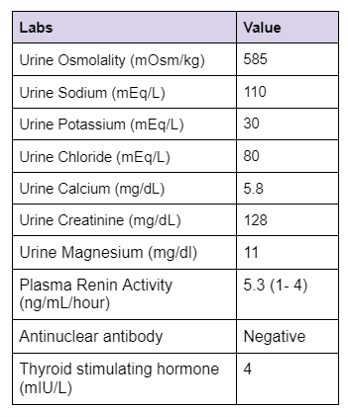
Kidney potassium excretion is regulated by serum potassium levels, distal sodium delivery and aldosterone. In the setting of hypokalemia the appropriate kidney response is to conserve potassium. If urinary potassium is high or inappropriately normal in the face of hypokalemia, it implies kidney potassium wasting.
A urine K/Cr ratio of >15 mEq/g (15 mmol/g or 1.7 mmol/mmol) suggests kidney losses of potassium. The ratio is expressed in different units and sometimes can be confusing.
A K/Cr ratio of 15 mEq/g equals 15 mmol/g (1 mEq of K = 1 mmol). Alternatively it can be expressed as mmol/mmol (1 g creatinine = 8.8 mmol).
So a K/Cr ratio of 15 mEq/g = (15 X 1)/ 8.8 mmol/mmol = 1.7 mmol/mmol.
The urine K/Cr ratio in our patient was 23 mEq/g (2.6 mmol/mmol) confirming urinary losses.
Step 3 Once the kidney is identified as the culprit of potassium loss, blood pressure and acid-base status help differentiate the causes of hypokalemia as shown in the algorithm-
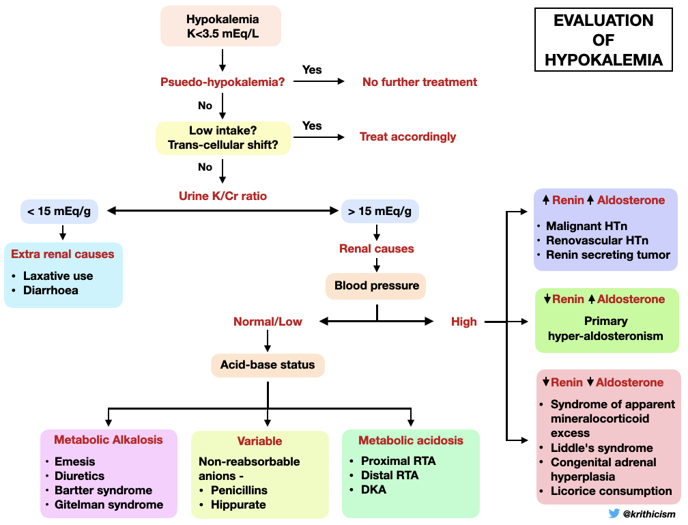
The presence of hypokalemia, normal/low blood pressure and metabolic alkalosis in our patient, narrowed down our diagnosis to either vomiting, diuretic use, Bartter or Gitelman syndrome.
Step 4 Look at urine chloride levels
Urine chloride excretion >10 rules out GI losses such as vomiting or nasogastric drainage.
Step 5 How do you differentiate Gitelman from Bartter syndrome? Answer- Calcium/ creatinine ratio
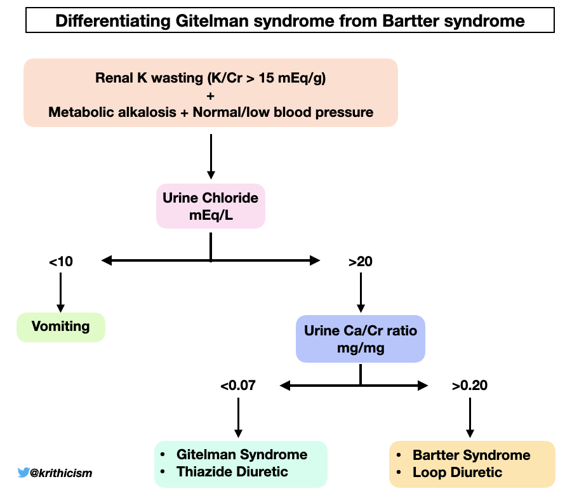
Calculating the urine Calcium-to-creatinine ratio in our patient-
Urine Ca/Cr (mg/mg) = 5.8 mg/dL / 128 mg/dL= 0.04 mg/mg (0.1 mmol/mmol)
A calcium/creatinine ratio <0.07 mg/mg (0.2 mmol/mmol) is seen in Gitelman syndrome and in patients using thiazide diuretics.
While converting Ca/Cr ratio from mg/mg to mmol/mmol, it’s important to note that 1 mg Ca = 0.025 mmol and 1 mg Creatinine = 0.0088 mmol
So a Ca/Cr ratio of 0.07 mg/mg = (0.07 X 0.025)/0.0088 = 0.2 mmol/mmol
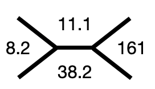
Step 6 Calculating the fractional excretion of magnesium and chloride


With the above evidence of urine calcium/creatinine ratio <0.07 mg/mg, increased fractional excretion of magnesium and chloride in a patient with hypokalemia, metabolic alkalosis, low/normal BP and no history of thiazide use, we narrowed down our diagnosis to Gitelman syndrome.
Why do we think it was Gitelman syndrome?
Gitelman Syndrome
Also called familial hypokalemia-hypomagnesemia is a rare salt-losing tubulopathy characterized by
- Hypokalemia
- Metabolic alkalosis
- Hypomagnesemia
- Hypocalciuria, and
- Normal/ low blood pressure.
Its prevalence is estimated to be between 1 to 10 in 40,000. Gitelman syndrome occurs as an autosomal recessive disorder due to inactivating mutations in the SLC12A3 gene that encodes the thiazide-sensitive sodium-chloride cotransporter (NCC) in the distal tubule. The Na-K-ATPase pump in the basolateral membrane transports sodium out of, and potassium into, the cell. The entry of sodium and chloride into the cell is mediated by NCC in the apical membrane which is inhibited by thiazide diuretics.
There are over 350 mutations identified throughout the SLC12A3 gene that can cause Gitelman syndrome. A Gitelman syndrome-like phenotype, including hypomagnesemia and hypocalciuria, is also associated with mutations in the CLCNKB gene encoding the chloride channel ClC-Kb, the cause of classic Bartter syndrome. The localization of ClC-Kb in the distal convoluted tubule explains the phenotypic overlap with Gitelman syndrome. Less frequently Gitelman syndrome can present with normal magnesium levels.
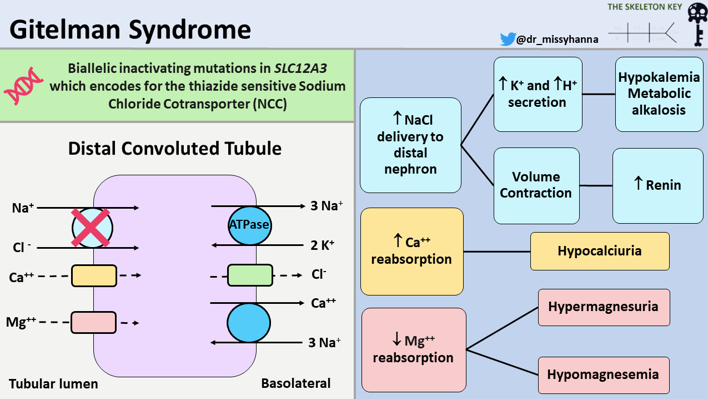
Figure 3. Pathophysiology of Gitelman syndrome
Reduced NCC activity mimics the action of thiazide diuretics causing
- Kidney potassium wasting and hypokalemia
- Kidney magnesium wasting and hypomagnesemia
- Volume contraction
- Low urine calcium excretion
- Increased renin and aldosterone levels
Patients with Gitelman syndrome usually present in late childhood or adulthood. Majority of patients have mild or nonspecific symptoms, although severe life-threatening complications have been described. The clinical manifestations include:
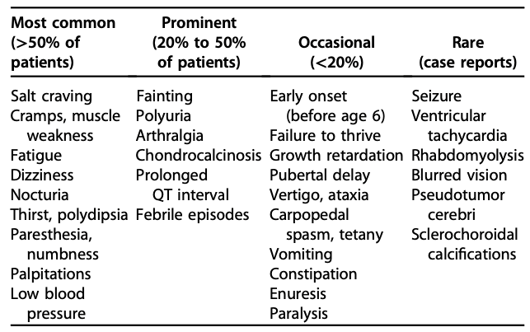
Chondrocalcinosis (calcification of the cartilage) and sclerochoroidal calcifications (calcification of sclera and choroid) are rare complications seen in Gitelman syndrome. This happens because low magnesium decreases the solubility of calcium pyrophosphate crystals thus promoting the deposition of these crystals. Ultrasound /X-ray should be performed if the patient has complaints suggestive of chondrocalcinosis and an ophthalmology exam is warranted when sclerochoroidal calcification is suspected. Prolonged QT interval, ventricular arrhythmias and sudden cardiac deaths, although rare, have been reported in patients with Gitelman syndrome. Therefore a baseline ECG is recommended for all patients.
Diagnostic criteria
The presence of both hypocalciuria and hypomagnesemia is highly predictive for the clinical diagnosis of Gitelman syndrome. But this may also be seen in classic Bartter syndrome, which makes the diagnosis difficult. Gitelman syndrome has a more benign course when compared to Bartter syndrome which is associated with nephrocalcinosis, proteinuria and rarely even progresses to kidney failure. The criteria to diagnose Gitelman syndrome includes clinical, biochemical parameters often supported by genetic testing.
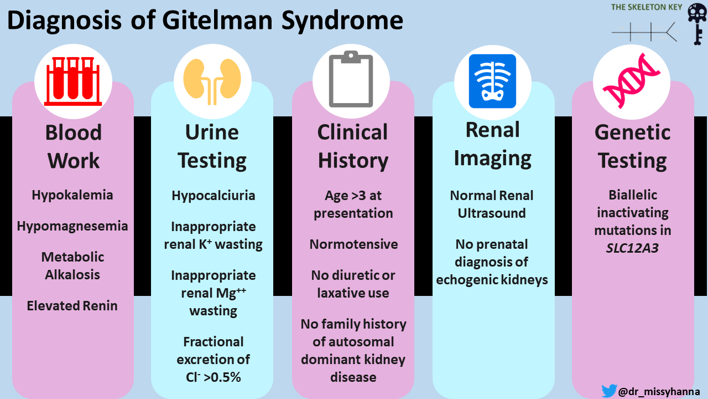
Recommended diagnostic criteria for Gitelman syndrome

Clinically suspected patients should be offered genetic testing in order to confirm the diagnosis of Gitelman syndrome.
Differential diagnosis
1. Diuretic use
2. Classical Bartter syndrome or type III – Occurs at a younger age, with failure to thrive, polyuria with usually normal serum magnesium levels.
3. HNF1B mutations – Present with hypomagnesemia and hypocalciuria as seen with Gitelman syndrome and are also associated with maturity-onset diabetes of the young, early chronic kidney disease, family history, abnormal liver enzymes, renal or urogenital malformations or kidney cysts
4. KCNJ10 mutations – Cause autosomal recessive EAST syndrome (epilepsy, ataxia, sensorineural deafness and tubulopathy)
Gitelman-like syndrome has also been described in association with autoimmune diseases and drugs such as cisplatin.
D. Confirmatory Testing and Final Diagnosis
Genetic analysis for many of the suspected gene mutations in Gitelman syndrome is available commercially. The practicality of genetic testing has been limited by the large size of the involved genes, the multitude of recognized mutations, intrafamilial heterogeneity, and cost.
The detection of biallelic inactivating SLC12A3 mutations is diagnostic of Gitelman syndrome. The analytical sensitivity and specificity of genetic testing is 90% to 100% and 100%, respectively. The clinical sensitivity (proportion of positive tests if the disease is present) is 65% to 80% depending on the tests used. Some individuals who have a clinical picture consistent with Gitelman syndrome may not have mutations detected in both SLC12A3 alleles (ie, identified as heterozygotes).
In our patient, features that were consistent with Gitelman syndrome were-
- Age of presentation
- Hypokalemia
- Metabolic alkalosis
- Normal blood pressure
- Hypomagnesemia
- Hypocalciuria
- FeCl >0.5 %
Genetic testing could not be done due to its cost and non-availability at our centre. Though still expensive, the cost of these tests continue to come down and may be more accessible in the near future.
E. Management
The first step in the management of Gitelman syndrome is to look for acute and severe manifestations of hypokalemia and hypomagnesemia such as cardiac arrhythmias, quadriparesis, respiratory weakness which warrants immediate IV replacement of magnesium and potassium. Since our patient presented with weakness and cramps, she was first treated with IV magnesium followed by IV potassium. In the presence of hypomagnesemia, magnesium should be replenished first. The “why” of this is covered in one of our previous cases.
Following improvement in her symptoms, she was switched over to oral potassium and magnesium salts. A target of at least 3.0 mEq/L (3 mmol/L) for potassium and 1.4 mg/dl (0.6 mmol/L) for magnesium is recommended (KDIGO consensus). Patients with Gitelman syndrome are followed up closely and regularly until stable levels of serum electrolytes are achieved after which at least an annual follow up is advocated.
Potassium replacement should ideally be given as chloride (KCl) as chloride is the main anion lost in the urine and patients are alkalotic. Magnesium should be replaced as organic salts (aspartate, citrate, lactate) as they have a higher bioavailability than their oxide and hydroxide salts. Magnesium replenishment is also the cornerstone in the prevention of chondrocalcinosis. Patients with Gitelman syndrome should be educated about the side effects of these drugs- such as pain in the abdomen, gastritis, diarrhea.
Besides correcting magnesium and potassium, patients with Gitelman syndrome are also encouraged to maintain liberal salt intake.
In patients having persistent hypokalemia despite adequate potassium and magnesium replacement, the use of potassium-sparing diuretics, RAAS inhibitors, and NSAIDS such as indomethacin has been proposed. The acute reduction of angiotensin II or aldosterone levels by these drugs can cause symptomatic hypotension and should be administered with caution (Blanchard A et al)
Caution must be taken in situations such as
- Pregnancy which aggravates hypokalemia and hypomagnesemia – appropriate supplementation and electrolyte monitoring should be advised.
- Anesthesia – Hypokalemia and hypomagnesemia which may potentiate the effects of anesthesia (such as neuromuscular blockade). Achieving a target of >3mmol/L for potassium and >0.5 mmol/L for magnesium is advised (NICE guidelines) to avoid such complications.
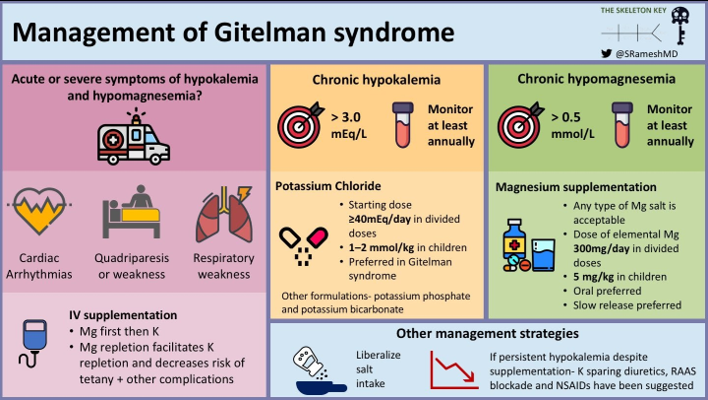
Long term management
Educating the patient and caregiver about the long term management and outcome is essential. Gitelman syndrome can affect school performance, work performance and quality of life (QOL). Patients were found to have particularly low QOL scores in terms of role limitations caused by physical health, emotion, level of energy, and general health perception. It was similar in range to that described for diseases such as hypertension, diabetes, congestive heart failure, and coronary artery disease and slightly worse with respect to role limitation due to physical health, emotion, and social function. Decreased QOL may be related to the cost of medications, the large number of pills required to maintain their electrolytes within reasonable range, the limited understanding of their disease, lack of good evidence regarding treatment options, doctors’ perception that it is benign, among other factors. These factors must be considered when treating Gitelman syndrome and measures to improve QOL should be developed on an individual basis.
F. Take Home Points
- The classical presentation of Gitelman syndrome is chronic hypokalemia with metabolic alkalosis, hypocalciuria, hypomagnesemia and low or normal blood pressure.
- A calcium/creatinine ratio <0.07 mg/mg is seen in Gitelman syndrome and a ratio >0.20 favors the diagnosis of Bartter syndrome.
- Gitelman has a more benign course than Bartter syndrome. It presents in late childhood or early adulthood whereas Bartter syndrome presents in infancy or early childhood.
- The presence of biallelic inactivating SLC12A3 mutations by genetic testing confirms the diagnosis but its absence does not exclude the diagnosis Gitelman syndrome.
- Potassium and magnesium replacement to maintain normal/near normal serum levels is the key to managing Gitelman syndrome.


Excelente caso!
Very helpful and detailed explanation. Thank you
Excellent case!