Washington University School of Medicine in St. Louis
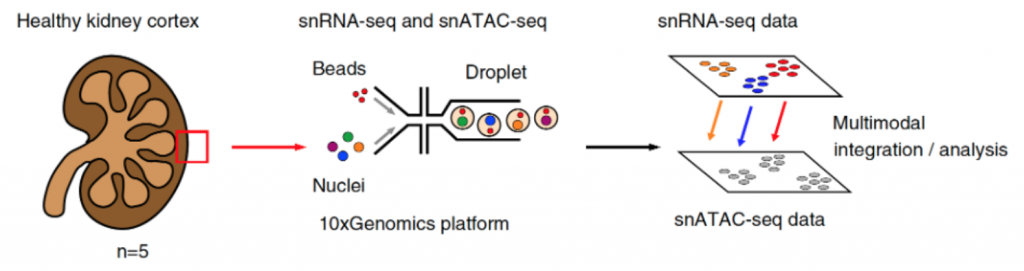
The reach of single cell sequencing technologies continues to rapidly expand across tissue and disease models with the introduction of multi-omic approaches. Here, we continue our conversation on sequencing applications by discussing the recent work of Muto et al, where single nucleus RNA (snRNAseq) and single nucleus ATAC (Assay for Transposase-Accessible Chromatin) sequencing (snATACseq) were paired to better define the characteristics of the heterogeneous cellular landscape that comprises the kidney.
So, how does the addition of snATACseq enhance the transcriptional atlases that have been previously generated with snRNAseq?
To understand this, we need to begin with the building blocks, namely chromatin. Chromatin is made up of basic units of histone proteins and DNA called nucleosomes. The timing and degree of chromatin opening (accessibility) determines which macromolecules and chromatin binding factors have the opportunity to bind to DNA, a process critical for organ development and cellular repair (Klemm et al, 2019; Muto et al, 2021). In essence, genes are packed away neatly within chromatin structures until they need to be accessed at which time they change to an open structure – much like needing, finding, and opening a file you need that was tucked away on your desktop. While it was originally thought that the chromatin accessibility of molecules would vary greatly, it has instead been reported that there is a great degree of evolutionary conservation (Klemm et al, 2019; Buenrostro et al, 2013). To determine cell type specific levels of accessibility and therefore the effect of chromatin modification on transcription, snATACseq uses Tn5 transposase, a hyperactive enzyme that can cut open chromatin. The resulting nicked chromatin is “tagged” with known sequencing adaptors — these “tags” allow fragments to be amplified and sequenced.
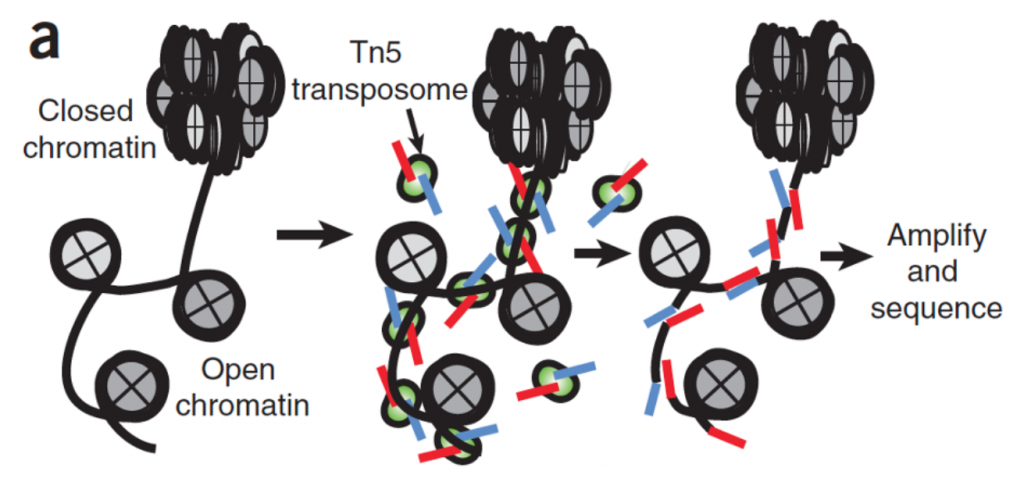
While snATAC-seq is not the only technology used for the measurement of open chromatin (you can read more about other modalities including NOMe-seq, DNase-seq, and MNase-seq from Klemm et al, 2019), it does have advantages. snATAC-seq requires a lower number of cells and has a relatively simple protocol for generation of high quality sequencing libraries. This makes snATAC-seq a great option for limited samples, such as clinical kidney biopsies or other human tissue samples. In their study, Muto et al sequenced five non-tumor kidney cortex nephrectomy samples (n = 3 males and 2 females), where all major cell types (proximal tubule, thick ascending limb, podocytes, etc) could be detected, as well as fibroblasts, leukocytes, and a subpopulation of VCAM1 and HAVCR1 positive cells. In the proximal tubule, VCAM1 and HAVCR1 have been associated with acute injury as well as long term consequences like failed repair (Schulz et al, 2020; Kirita et al, 2020). Furthermore, in their analysis, snATAC-seq enabled the delineation of the proximal convoluted tubules (PCT) and proximal straight tubules (PST) by chromatin accessibility of SLC5A2 (SGLT2) and SLC5A1 (SGLT1), respectively. This effectively demonstrated a differentiation of S1-2 and S3 segments in the proximal tubule that was more difficult to resolve with snRNAseq data alone.
Additionally, the increased sensitivity of snATAC-seq aids in the identification of differentially accessible chromatin regions (DARs), which help determine changes in regulatory element activity across biological and/or pathophysiological conditions (Gontarz et al, 2020). DARs can be found most commonly near transcriptional start sites and adjacent promoter regions, but can also be found in intronic regions (Muto et al, 2021). Once DARs have been identified, the presence of certain binding motifs can indicate predicted activity of transcription factors and taken further, this can culminate in the definition of cell-type-specific transcription factors that affect chromatin accessibility. Patterns, or “families”, of chromatin regions that alter accessibility together, are called cis-coaccessibility networks (CCAN) and can be used to predict potential chromatin interactions across the kidney (Muto et al, 2021).
Despite all the bioinformatic advantages of multi-omic approaches, the importance of validation is still front and center. One strategy for validation of snATACseq is the employment of chromatin immunoprecipitation with quantitative PCR (ChIP-qPCR) – this technique quantifies DNA expression in cultured cells or tissues. For validation, Muto et al used primary renal proximal tubule epithelial cells (RPTECs) to investigate HNF4A binding that is critical for proximal tubule differentiation. However, this approach can create roadblocks for validation studies if relevant cell lines are not reliable or available. Alternative approaches to validating findings from multi-modal analyses include dCas9-DNMT1 or -KRAB, which allow investigators to change accessibility of chromatin and observe downstream changes in gene expression (Muto et al, 2021).
As the rapid wave of sequencing technologies continues to expand, it is critical to keep questions relevant to kidney physiology in mind and determine how these different modalities help us get closer to defining the cellular landscape of the kidney. Muto et al present a resource for pairing snRNA-seq and snATAC-seq to closely investigate heterogeneity in the proximal tubule, as well as the thick ascending limb. In the future, this work will be expanded to look at changes in chromatin accessibility and rare cell types across disease states, so that we may better understand the pathobiology of the kidney.
Reviewed by: Elinor Mannon, Kelly Hyndman, PhD, and Matthew Sparks, MD
If you enjoyed reading this post and want to learn more about RNA-sequencing and its use in kidney research, consider reading this post:

I found this article very interesting, thanks for sharing