Post by: Dearbhla Kelly, Dublin, Ireland
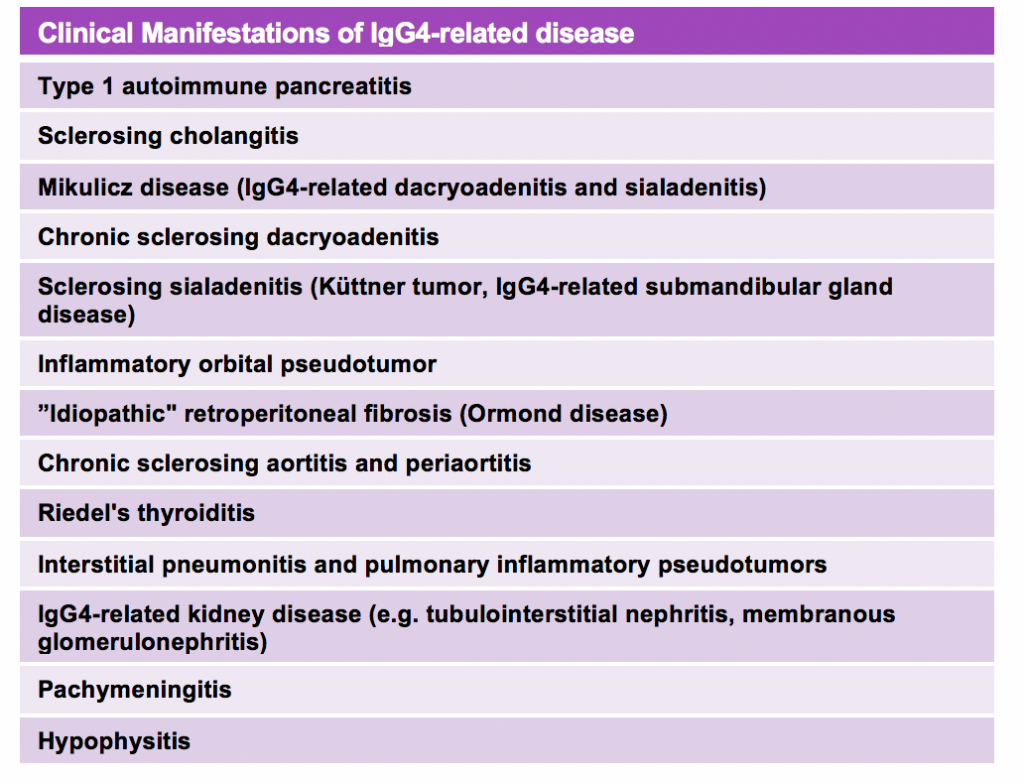
Every so often in the Nephrology clinic, I come across this uncommon, but increasingly recognized entity of IgG4-related disease (IgG4-RD). IgG4-RD is an immune-mediated, fibrous and chronic inflammatory condition that comprises a collection of disorders that share specific pathologic, serologic, and clinical features. The pathologic hallmark of IgG4-RD is dense lymphoplasmacytic infiltration with a predominance of IgG4-positive plasma cells in the affected tissue. Serum IgG4 levels are typically elevated in approximately two thirds of patients but these antibodies are not thought to be pathogenic themselves. The pathogenesis of IgG4-RD is poorly understood but appears to be autoimmune with an important role for T cells, especially CD4+ and T-follicular helper cells. It most commonly affects middle-aged and older men, but can also be seen in women. Clinical manifestations of disease are summarized in Table 1.
Four patterns of renal involvement have been described:
1. Tubulointerstitial involvement
2. Glomerular involvement
3. Retroperitoneal fibrosis
4. Overlap with ANCA-associated vasculitis (AAV)
Tubulointerstitial nephritis (TIN) is the most common IgG4-related kidney disease. The pathologic hallmark is the presence of a diffuse or multifocal lymphocytic infiltrate with a predominance of IgG4+ plasma cells, an IgG4/IgG-positive plasma cell ratio >40% and >10 IgG4-positive plasma cell per high-power field (Figure 1). Tubulitis is usually present, with mononuclear cells, plasma cells and eosinophils. Granular deposits of IgG and C3 can be seen on the tubular basal membrane with immunofluorescence (IF). Two proposed criteria for the pathologic diagnosis of IgG4-TIN have been proposed by the Mayo Clinic and the Japanese Society of Nephrology. Hypocomplementemia is present in 60% of patients. TIN can cause an AKI or CKD in this context with variable levels of haematuria or proteinuria. Urinary white blood cells or casts are not always present in these cases.
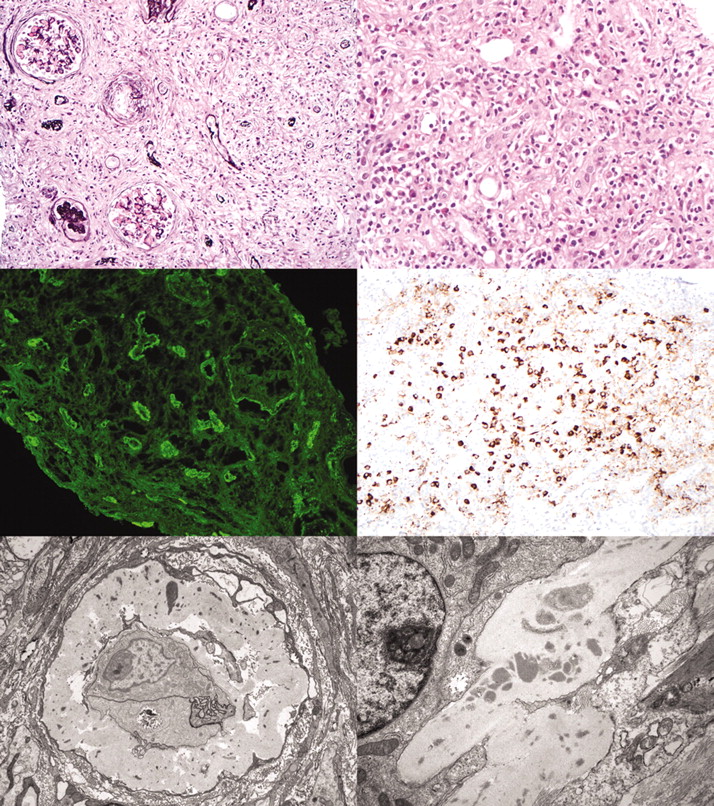
Figure 1: Typical pathologic features of IgG4-related TIN. Top, left: An expansile, destructive fibroinflammatory process with residual tubular basement, membranes seen on a silver stain. Glomeruli are unaffected except for periglomerular fibrosis and global glomerulosclerosis (Jones-methenamine silver, ×200). Top right: The interstitial infiltrate is composed of mononuclear cells, plasma cells, and several eosinophils. Mononuclear cell tubulitis is seen focally (hematoxylin and eosin, ×400). Middle left: Immunofluorescence for IgG shows diffuse granular tubular basement membrane staining, as well as Bowman’s capsule staining. Middle right: Immunohistochemical staining for IgG4 shows a marked increase in IgG4-positive plasma cells in the infiltrate (×200). Bottom left: Electron microscopy reveals thickened tubular basement membranes with scattered electron dense immune complex deposits in an atrophic tubule (×5800). Bottom right: Amorphous tubular basement membrane deposits (×9700). Reproduced from JASN July 2011, 22 (7) 1343-1352
Membranous nephropathy is the most frequent form of glomerular involvement, occurring in approximately 7% of these patients. Rarely, IgA nephropathy and membranoproliferative glomerulonephritis have also been described. Glomerular disease usually coexists with TIN but can occur in the absence of tubular involvement. Nephrotic range proteinuria is the most common presentation. IF shows IgG and C3 deposits in glomeruli and abundant electron dense deposits in the subepithelial space are visible by electron microscopy.
Idiopathic retroperitoneal fibrosis (RPF) is a rare fibro-inflammatory disease of uncertain aetiology characterized by periaortic and peri-iliac fibrosis. Over 50% of cases are thought to be attributable to IgG4-RD and sometimes it is the only disease manifestation. RPF can entrap both ureters causing obstructive uropathy which can present with lower back and flank dull pain as well as non-specific constitutional symptoms such as anorexia, weight loss, fatigue.
Overlap with ANCA-associated vasculitis has also been described though the exact incidence of AAV-IgG4-RD overlap is unknown. In this study, most cases were associated with the granulomatosis with polyangiitis subtype. The IgG4-RD manifestations reported in this context were chronic peri-aortitis, orbital mass and tubulointerstitial nephritis, prevertebral fibrosis, pachymeningitis and autoimmune pancreatitis. Both AAV manifestations and IgG4-RD lesions can relapse.
IgG4-RD diagnosis is usually confirmed by biopsy of the involved organ in an otherwise appropriate clinical or radiological context. Further investigations should include a CT of the thorax, abdomen, and pelvis, a urinalysis, serum complement and immunoglobulin levels, HbA1c, CBC with differential, renal and liver function testing, amylase and lipase. PET scanning can also be useful to determine the disease extent. To evaluate retroperitoneal lesions, contrast-enhanced CT scan, MRI, or PET/CT are the preferred tools though ultrasound will detect any hydronephrosis present.
In terms of treatment, glucocorticoids are generally recommended as first-line remission induction agents (typically 30-40 mg daily). Most patients tend to respond to glucocorticoid therapy within weeks as evident from symptomatic or organ function improvement, reduction in the size of masses/organomegaly, or a decrease in serum IgG4 levels. Patients with established CKD, even including those with advanced disease, can still demonstrate some limited improvement after steroid treatment, but they are at risk of developing renal cortical atrophy. In those who don’t respond, can’t be tapered, or have contraindications to glucocorticoids, rituximab (1 g IV every 2 weeks for a total of two doses) is suggested as next in line or as an early glucocorticoid-sparing approach. In an open-label trial, 97% of patients responded to Rituximab, with 40% remaining in complete remission at 1 year. If rituximab is unavailable, alternative agents would include azathioprine or mycophenolate mofetil. However, these appear to not be as effective as Rituximab with one series suggesting that nearly half of all patients will relapse on these immunomodulators. Patients with organ-threatening IgG4-RD manifestations and those with an elevated risk of relapse (e.g. multiorgan disease, significantly elevated serum IgG4, involvement of the proximal bile ducts, or a history of prior relapse) will likely benefit from maintenance therapy with steroid-sparing agents to minimize morbidity. In the case of RPF, relief of urinary obstruction may also be necessary via placement of ureteral stents, percutaneous nephrostomy or open surgery (which may also facilitate biopsy).
Here are some links to earlier RFN posts on this interesting topic:
IgG4 related kidney disease: A new disease or an old disease raising its head?
IgG-4 associated kidney disease
Post reviewed by Matthew A. Sparks
i found this article very interesting and important, it has a lot of info i need to know, thanks for sharing this info