Welcome to the 22nd case of the Skeleton Key Group, a team of 50-odd nephrology fellows who work together to build a monthly education package for the Renal Fellow Network. The cases are actual cases (without patient identifying information) that intrigued the treating fellow.
Written by: Tomas Guerrero
Visual Abstract: Deep Phachu
A. The Stem
A 63-year-old man with metastatic pancreatic neuroendocrine cancer presents to the hospital with severe and persistent vomiting, 8-10 times a day, for the past week and 1-2 episodes of diarrhea a day that had been less responsive to his home loperamide. This was associated with reduced oral intake, thirst, worsening fatigue, and numerous falls. Previous work up for recurrent episodes of nausea, vomiting, and diarrhea led his outpatient to diagnose his cancer and associated carcinoid syndrome.
Pancreatic neuroendocrine cancer was diagnosed 20 years ago with numerous metastatic nodules in the mesentery and liver (Rx with resection of liver lesions). He started on sandostatin 5 years ago after having more liver lesions and symptoms concerning for carcinoid and increased 5-Hydroxyindoleacetic acid (5-HIAA) levels. Liver lesions were ablated. Sandostatin was replaced by peptide receptor radionucleotide therapy (lutetium Lu 177 dotatate) 4 years prior with control of symptoms (diarrhea).
Findings on a computed tomography (CT) abdominal scan one month prior to admission showed bowel edema and ascites. The ascites was described as being significantly increased throughout the abdomen, with a 6.9 x 4.7 cm mass in the mesentery, that was stable as compared to previous scans. However, what was new in this scan was the development of diffuse thickening of the small bowel and colon consistent with bowel wall edema. He had been started on spironolactone and furosemide in response to the aforementioned changes, but he had discontinued the diuretics two days prior to his admission to the hospital.
His other home medications include haloperidol, lactulose, loperamide as needed for excessive diarrhea, mirtazapine, olanzapine, tamsulosin, cetirizine, pantoprazole, clonazepam as needed for anxiety, and trazodone as needed for insomnia.
Physical Examination:
BP 121/82 mm Hg, heart rate 75 bpm, respiratory rate 12/ min, Temp 98.1° F, SpO2 97% on room air. Weight 72.6 kg.
He was awake, but appeared confused and lethargic. He had dry mucous membranes, cool extremities with poor skin turgor. Neurologic examination showed mild weakness on all extremities with no sensory deficits. His pulmonary, cardiac and abdominal exam were normal.
B. The Labs
His labs 1 month prior to the presentation:
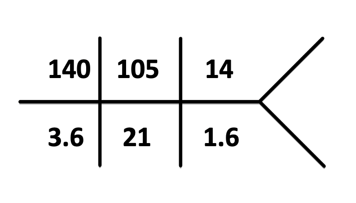
Labs on admission:
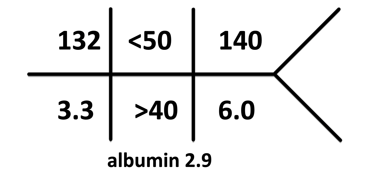
ABG: pH 7.55, HCO3 60, pCO2 69, pO2 108
Labs show hyponatremia, hypokalemia, severe hypochloremia, metabolic alkalosis, azotemia, and acute kidney Injury (AKI).
At the time of his consult, he had no urine available for microscopy or urine electrolyte evaluation.
Acid-base derangement interpretation
Blood gas shows metabolic alkalosis, respiratory acidosis and an acidemia that was interpreted as an anion gap metabolic acidosis. The expected respiratory compensation for a metabolic alkalosis predicts that the pCO2 should be around 62 土 2. The corrected anion gap, when taking albumin into account, is at least 26.
C. Differential Diagnoses and More Data
Nephrology was consulted for the severe metabolic derangements and AKI. Let’s break down what is going on, as we have the following:
- Hypovolemic hyponatremia – By exam, the patient looked hypovolemic given his dry skin, dry mucous membranes, and feeling of thirst. The hypovolemia was brought about by severe vomiting in the setting of concomitant loop diuretic use. His hypovolemia is likely leading to appropriate renin-angiotensin-aldosterone system (RAAS) activation as a means to correct the hypovolemia. Moreover, anti-diuretic hormone (ADH) is likely appropriately increased, especially in the setting of nausea and vomiting leading to a high urine osmolarity, although urine was unavailable in the initial evaluation. However, in this individual the hyponatremia is quite mild.
- Anion gap metabolic acidosis – The presumed cause of his anion gap (26) was metabolic acidosis was the accumulation of organic anions due to impaired clearance from severe AKI. The mnemonic GOLDMARK can be used in the differential for an anion gap metabolic acidosis, and can be found detailed in our previous case.
- Hypokalemia – Interestingly, gastric content (vomitus) does not contain much potassium (only 10 mEq/L). Thus, hypokalemia is likely due to a combination of a highly activated RAAS, bicarbonaturia, and furosemide. Secondary hyperaldosteronism leads to the activation of epithelial sodium channel (ENaC) and Na+/K– ATPase in the principal cells of the collecting duct allowing K+ secretion down a favorable concentration gradient. Increased distal delivery of Na+ and HCO3– also leads to increased KHCO3 excretion.
- Severe hypochloremia – The most striking finding on the labs is his hypochloremia. This is due to gastric losses of Cl– (along with H+) during multiple episodes of vomiting. More on this below.
- Metabolic alkalosis – While there are many mechanisms to the development of metabolic alkalosis, in this case, it was the combination of emesis and diuretic use.
Gastric losses (also discussed extensively in one of our earlier SKG cases): The gastric generation of bicarbonate can be separated into a generation and maintenance phase:
Bicarbonate generation phase: HCl secretion into the gastric lumen generates an equimolar amount of HCO3– to the extracellular fluid (ECF).
- normal conditions– H+ enters the duodenum and is neutralized by HCO3– (secreted by the pancreas) to generate CO2 and water.
- During vomiting– H+ (and an equimolar amount of Cl–) is removed from the body (because of the vomiting), HCO3– generated in the pancreas is not used for neutralization, and is instead added to the extracellular fluid (ECF) causing metabolic alkalosis.
Maintenance phase (of ↑HCO3–):
- normal conditions- As HCO3– is added to the ECF, the kidneys initially compensate by removing the excess HCO3– as NaHCO3 by filtration and excretion.
- During vomiting- The continued loss of gastric fluids via vomiting (particularly Cl– loss) and concomitant kidney loss of NaHCO3 leads to even further ECF contraction. This leads to a propensity to increase proximal and distal HCO3– reabsorption. Moreover, ECF contraction conspires to eventually cause diminished GFR in an attempt to maintain euvolemia, also decreasing HCO3– filtration.
Lastly, hypokalemia contributes to the maintenance of metabolic alkalosis by:
- stimulating NH4+ generation and excretion (and increasing bicarbonate generation) in the proximal tubule,
- increasing H+ secretion in type A intercalated cells,
- limiting bicarbonate secretion in the type B intercalated cells
Diuretic induced alkalosis:
Furosemide inhibits the Na+-K+-2Cl– cotransporter in the thick ascending limb of the loop of Henle, increasing delivery of Na+ to the distal tubule. Together with secondary hyperaldosteronism, this leads to increased distal Na+ reabsorption, K+ secretion, and H+ secretion, contributing to metabolic alkalosis and hypokalemia.
D. Final Diagnosis & Management
When the team was originally consulted, the first question was whether or not this patient needed dialysis. The severity of AKI, clinical deterioration, metabolic derangements, confusion, all painted a picture of severe kidney failure leading to uremia. All of the aforementioned findings can be explained by electrolyte losses, either by emesis or by urine. While he was anuric at the time of consultation, it is important to consider his history of severe vomiting and low oral intake, and start with adequate volume resuscitation first, before considering any plans for dialysis.
This type of metabolic alkalosis is repaired by adequate Cl– repletion. It is also necessary to manage the underlying cause —stop gastric losses of HCl.
There are several options for chloride solutions that can be used to replete the volume and correct the metabolic alkalosis: sodium chloride (NaCl), potassium chloride (KCl) or hydrochloric acid (HCl). The use of intravenous IV HCl can be considered in patients unable to receive volume resuscitation, but proper preparation and storage can be tedious. A retrospective analysis of IV HCl administration in 30 patients with diuretic-induced metabolic alkalosis, resulted in correction of hyper bicarbonatemia and hypochloremia with no apparent adverse events. If IV HCl is attempted, the patient should be monitored closely in an ICU setting.
Our patient was given continuous IV saline/NaCl hydration the next few days, while monitoring serum chloride, potassium, bicarbonate and creatinine. In this case, a fluid rich in both sodium (154 mEq/L) and chloride (154 mEq/L) is preferred over balanced solution. Moreover, there is no need to give more bicarbonate (or bicarb precursor- lactate) which is contained in balanced solutions. Repletion of chloride with normal saline (1) restores extracellular volume, normalizes the GFR, improves the urine output, and (2) corrects the metabolic alkalosis by decreasing proximal HCO3– absorption and increasing HCO3– secretion in the distal nephron. The correction of bicarbonate and chloride during the hospital stay can be seen in the figure below:
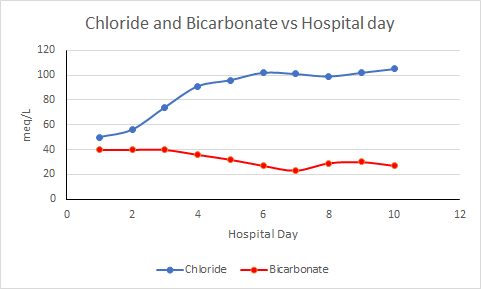
By his 6th day of hospitalization, IV fluids were discontinued, urine output improved to over a liter per day and serum creatinine was back to baseline at 1.7 mg/dL.
E. Take Home Points
- Secretion of HCl into the gastric lumen generates an equal amount of HCO3. During vomiting, this HCO3– is added to the ECF as Cl– is removed from the body.
- Hypochloremic metabolic alkalosis can be divided in 2 phases: bicarbonate generation (via loss of gastric & renal H+) and maintenance of increased serum bicarbonate levels.
- Loop diuretics can affect renal handling of electrolytes, which can lead to increased bicarbonate absorption.
- Hypochloremic metabolic alkalosis is repaired by chloride repletion. In circumstances that are not responsive to traditional chloride rich fluids, an infusion of hydrochloric acid can be considered to both reduce serum pH and increase serum chloride.
Reviewed and edited by Joel Topf, Sayna Norouzi, Dominique Tomacruz, Chi Chu, Alejandro Meraz, Kartik Kalra, Nasim Wiegley, Sudha Mannemuddhu, Jamie Willows, Matthew A. Sparks

This is a great case to discuss hypochloremic metabolic alkalosis. I would like to add that part of the mechanism is a protein Pendrin which is a chloride/bicarb exchanger in the collecting tubule. When patients are chloride depleted due to vomiting or diuretic use they are unable to exchange chloride for bicarb and bicarb accumulates in the blood.