Meloney Oliveira, MD
1st year fellow
Division of Nephrology
Rush University Medical Center
*The patient history in this post is fictitious, but represents how a patient with these biopsy findings might present
Case*: A 40-year-old with a 16-year history of heroin addiction administered through “skin popping” (the act of subcutaneous drug injection) presented with a 6 week history of lower extremity swelling. Physical examination revealed multiple fibrotic and flocculent nodules and crater ulcerations on the skin of his extremities where he had been injecting. The serum creatinine was 0.9 mg/dL and the albumin was 1.1 g/dL. The urinalysis showed 4+ protein and 2+ blood on urinalysis with 5 RBCs/hpf on microscopic analysis. The urine albumin to creatinine ratio was 4.2 g/g and the urine protein to creatinine ratio of 7.8 g/g. A kidney biopsy was performed and was consistent with secondary amyloidosis.
Background
Amyloidosis is a disorder of increased protein production, aberrant cleavage, misfolding and aggregation. Proteins that are usually soluble instead become insoluble and are pathologically deposited in the extracellular tissues as abnormal β-sheet structures that aggregate into fibrils. Accumulation of these deposits in the body organs leads to organ dysfunction and eventually death. There are different types of amyloidosis based on the origin of the misfolded proteins (Table 1). Here, we will be covering secondary amyloidosis which in this case is related to the deposition of serum amyloid A (SAA) protein as a result of chronic inflammation secondary to recurrent skin infections from “skin-popping” heroin.
Table 1: Common Subtypes of Amyloidosis.
Adapted from Benson et al – Simplified table of some of the most common amyloid fibrils subtypes.
| Disease Subtype | Protein Deposited | Disease Etiology |
| AL Amyloidosis (Primary) | Monoclonal immunoglobulin light chain | Plasma cell dyscrasias |
| AA Amyloidosis (Secondary) | Serum Amyloid A | Chronic inflammation or infection (See Table 2) |
| ATTR Amyloidosis | Mutant transthyretin Wild type transthyretin | HereditaryAge-related, senile ATTR |
| Aβ2M Amyloidosis | β2-microglobulin | Dialysis-related |
| ALECT1 Amyloidosis | Leukocyte chemotactic factor-2 | Unknown |
| AFib Amyloidosis | Mutant fibrinogen A | Hereditary |
| ApoA1 Amyloidosis | Apolipoprotein 1 | Hereditary |
| ALys Amyloidosis | Lysozyme | Hereditary |
Etiology and Clinical Classification
SAA protein is an apolipoprotein acute-phase reactant produced by the liver under the transcriptional regulation of proinflammatory cytokines. SAA is small (104 amino acids) and it has a three dimensional monomeric structure. Its role in the acute phase reaction remains poorly understood. In states of sustained chronic inflammation and chronic infection (Table 1) large quantities of SAA are produced. SAA is cleaved, misfolded and can aggregate into a highly ordered abnormal beta-sheet conformation known as an AA amyloid fibril. These amyloid fibrils combine with non-fibrillary constituents such as glycosaminoglycans (GAGs) and serum amyloid P component (SAP), to form deposits that disrupt the structure and function of tissues and organs. The amyloid that is seen related to SAA is not unique histopathologically. It demonstrates typical amyloid congo red affinity and has organized fibrils that are 8-12 nM in diameter on electron microscopy.
Table 2: Common Disorders Associated with AA Amyloidosis
| Chronic Diseases | Rheumatoid Arthritis Familial Mediterranean Fever Juvenile Idiopathic Arthritis Still Disease Ankylosing Spondylitis Psoriasis Behcet Syndrome Inflammatory Bowel Disease Vasculitis Castleman Disease Cryopyrin-associated Periodic Syndrome (CAPS) TNF-receptor associated Periodic Fever Syndrome |
| Infections | Leprosy Osteomyelitis Tuberculosis Chronic bronchiectasis Injection-drug abuse |
| Malignancy | Hodgkin’s Lymphoma Non-Hodgkin’s Lymphoma Renal Cell Carcinoma GI malignancy Lung Cancer Urogenital Cancer Mesothelioma |
Clinical manifestations and diagnosis
Many chronic inflammatory conditions have been associated with AA amyloid. In our case it was related to a long history of heroin use through “skin-popping”. This association was first described by Meador et al. in 1979. The intradermal injection of a foreign material into the skin induces local inflammation and a cellular reaction which predisposes the patient to poor wound healing, ulcers and frequent skin infections (i.e. cellulitis, abscesses, ulcerations). In the northwestern United States AA amyloidosis has been reported to occur not uncommonly in chronic heroin users that either skin pop or inject heroin intramuscularly and is thought to be related to a highly contaminated form of heroin that is available in that geographic area nicknamed “black tar heroin” because of its black color. As a result of the sustained chronic inflammation mediated by both infection and foreign body reactions, there is a constitutive production of SAA and for unknown reasons, certain patients develop an accumulation of amyloid. Laboratory testing for monoclonal immunoglobulins are useful in excluding AL amyloidosis and are usually negative in cases of AA amyloidosis. The kidney is the organ most frequently affected in AA amyloidosis. Initially, amyloid deposition in the kidney typically presents as proteinuria which can range from subnephrotic to massive (up to 20-30 g/day). Hypoalbuminemia may be profound and edema severe. Over time renal amyloid will often lead to progressive deterioration of kidney function and end-stage kidney disease.
The diagnosis of amyloidosis is made by biopsy with the pathognomonic Congo red positive staining with apple green birefringence under polarized light (See Figure 1). In order to subtype the culprit misfolded protein immunohistochemical staining can be used and in our case, immunohistochemical staining for SAA (Figure 1). Laser microdissection and mass spectrometry-based proteomic analysis can also be used to subtype the misfolded protein responsible for the amyloid deposits.
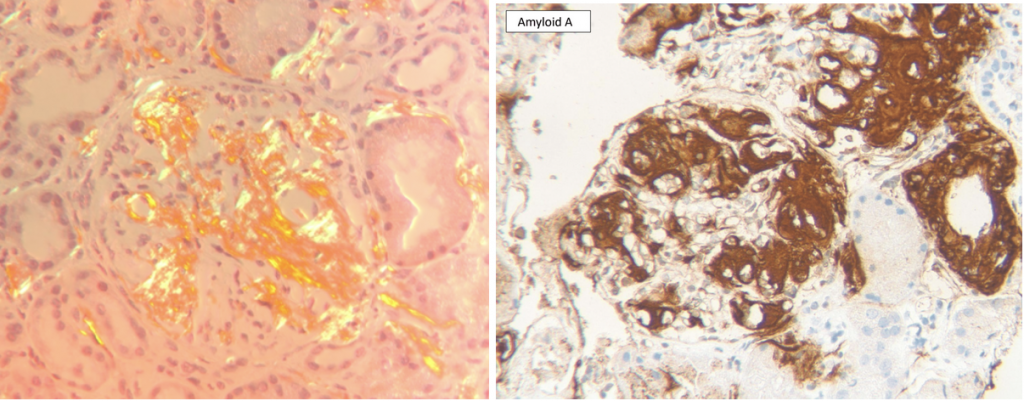
Images by Dr David Cimbaluk, Rush University Medical Center Renal Pathologist.
Other characteristic findings of kidney amyloidosis include the following:
Light microscopy:
Mesangial deposits are eosinophilic, acellular, amorphous, pale pink (classically weakly PAS positive), cotton candy-like material (Figure 2) as opposed to the deposits seen in diabetes which stain quite prominently on PAS. Glomerular basement membrane deposits are occasionally seen on Jones (silver) stain which are described as segmental subepithelial deposits that appear like the spikes of membranous nephropathy but tend to be longer and feathery, giving a “cockscomb” appearance (Figure 2). Arterioles and arteries can show amyloid deposits. Interstitial and tubular basement membrane amyloid deposits have similar pale pink, acellular appearances.
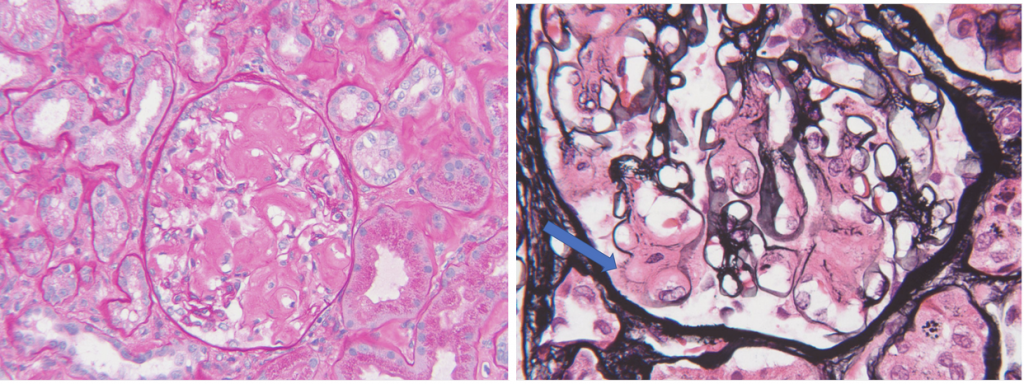
Images by Dr David Cimbaluk, Rush University Medical Center Renal Pathologist.
Electron microscopy:
Amyloid fibrils are extracellular, straight, randomly arranged (“hay stack”) 8-12 nM in diameter (Figure 3). Deposits are predominantly in the mesangium and glomerular basement membrane with typical projections extending towards podocytes with extensive foot process effacement.
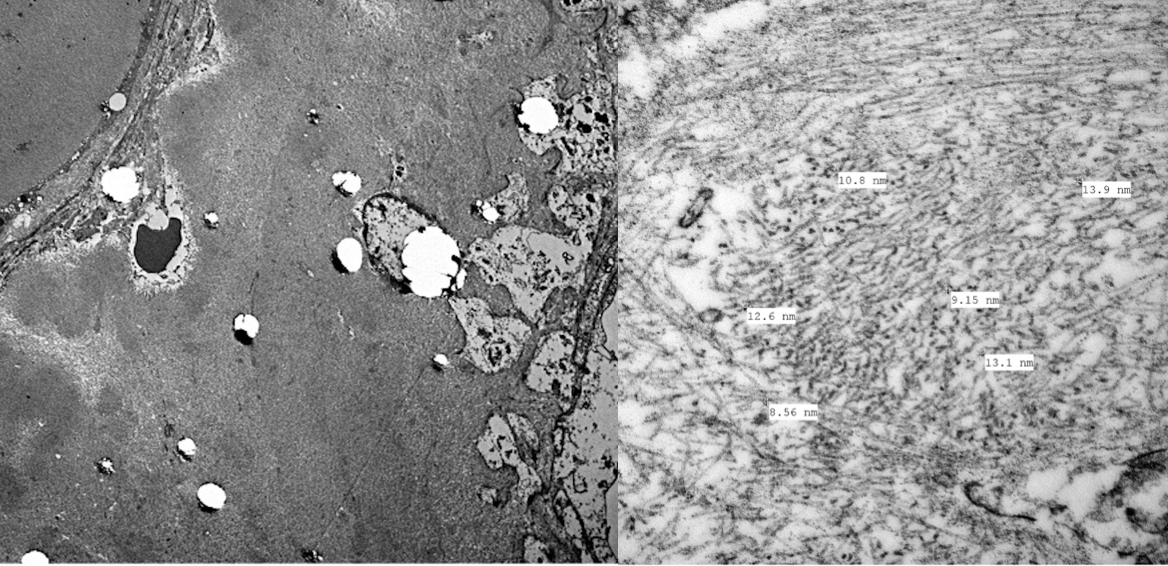
Images by Dr David Cimbaluk, Rush University Medical Center Renal Pathologist.
Treatment:
The focus of the treatment of AA amyloidosis is to inhibit the production of SAA. In this particular case, that would include extensive counseling of our patient on the importance of abstaining from drug use and offering a referral to a drug medication-assisted treatment center. If the patient is no longer subcutaneously injecting heroin, the nidus for chronic infections will be removed. SAA levels will normalize over time and there will no longer be a substrate for further amyloid production. In 1977, Dikman et al reported the resolution of proteinuria in patients with AA amyloidosis-associated nephrotic syndrome when the underlying inflammatory disease was in remission and SAA concentrations had returned to normal, despite the persistence of glomerular amyloid deposits. In 1989, Crowley et al reported a case of clinical improvement of heroin-associated renal amyloidosis after cessation of drug use.
Moreover, SAA is stimulated by inflammation-associated cytokines (i.e. interleukin (IL)-6, TNR and IL-1). Theoretically by inhibiting these molecules, SAA production would decline and lead to a decrease in amyloid production. Lane et al. used tocilizumab (an IL-6 inhibitor) in 20 patients with treatment-refractory AA amyloidosis and reported regression in amyloid deposition. Long term prospective studies are still needed to explore the efficacy and safety of this treatment.
Other agents have been developed in an attempt to treat AA amyloidosis by decreasing amyloid fibril polymerization. For example, eprodisate (a low molecular-weight molecule similar to heparan sulfate) works by binding competitively to GAG union sites and inhibits polymerization of amyloid fibrils. This prevents the stabilization of amyloid deposits, theoretically leaving the amyloid susceptible to proteolysis. A multicenter, randomized, double-blind, placebo-controlled trial in patients with AA amyloidosis-associated renal disease was conducted by Dember et al. The group reported that eprodisate reduced the progression of renal disease but it did not affect proteinuria or decrease the production of SAA. Unfortunately, this treatment option did not advance any further.
Richard et al targeted the SAP component of amyloid in another therapeutic strategy. It is important to recall SAP is responsible for the inherent resistance of amyloid proteolysis. They used (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine-2-carboxylic acid (CPHPC) to deplete SAP from the plasma. They then targeted the SAP that was in the amyloid deposits by using a humanized monoclonal IgG1 anti-SAP antibody. The binding of anti-SAP antibody to SAP in amyloid activates complement and attracts macrophages to engulf and destroy the targeted deposits. In 2018, Richard et al reported that repeat doses of this double strategy progressively removed amyloid from the kidneys, liver and spleen. SAP is present in all subtypes of human amyloid and therefore if the future trial data reports clinically significant benefits on morbidity and mortality, this treatment offers great promise for patients with all subtypes of amyloidosis.
The median survival after diagnosis depends on the primary disease and renal involvement is a worse prognostic factor.
Edited by: Pravir Baxi, MD, Roger Rodby, MD, Dearbhla Kelly, MD and Matthew A. Sparks, MD

Interesante caso clínico, gracias a Dios poco común